INTRODUCTION
Hearing loss (HL) is one of the most common diseases of all human birth defects, which can cause both hearing and speech disorder. Studies reported that 1%–3% newborns are diagnosed as congenital HL [1,2]. Among them, seventy percent is accounted for nonsyndromic hearing loss (NSHL; hearing loss as the only symptom). Hereditary HL can be inherited in an autosomal dominant, autosomal recessive, or X-linked recessive manner, as well as by mitochondrial inheritance [3,4]. According to the Hereditary Hearing Loss Homepage, more than 100 genes and 160 genetic loci have been described for NSHL (http://hereditaryhearingloss.org). The occurrence of mutations in GJB2, SLC26A4 genes, mitochondrial 12S rRNA and GJB3 were reported the most common genetic cause of NSHL [5,6]. In Beijing, nine hotspot mutations of these four genes have been screened in all newborns as a public health initiative since 2012 [7].
The solute carrier family 26 member 4 (SLC26A4), also named PDS gene, locates on chromosome 7q22.3-7q31.1 (NM_000441.1), encoding an anion transporter Pendrin protein. By acting as a chloride/bicarbonate transporter, Pendrin contributes to the homeostasis of ion concentration of the endolymph in the inner ear. It is believed that mutations of SLC26A4 are associated with enlarged vestibular aqueduct (EVA), the most common inner ear malformation in patients with NSHL [8,9]. The SLC26A4 mutational patterns include missense mutations, frameshift mutations, splice site mutations and insertions/deletions. Up to date, approximately 200 mutations in the SLC26A4 gene have been identified [10]. However, the mutation spectra in the coding exons of SLC26A4 are distinct in different populations and regions. It was found that 97.9% patients with EVA were detected with SLC26A4 mutations in Chinese, while only 40% in Caucasian populations [11,12]. Although SLC26A4 mutations are estimated to be the second most common genetic cause of human deafness, the precise molecular mechanisms underlying Pendrin function remain largely unknown.
In the present study, SLC26A4 gene was screened in two Chinese probands with EVA, as well as their normal family members. We identified two compound heterozygous mutations respectively (c.1174A>T/c.1181delTCT; c.919-2A>G/c.1023insC) in SLC26A4, which might affect Pendrin protein spatial structure. In addition, our study was the first time to identify the co-occurrence of c.1174A>T and c.1181delTCT in one patient. Notably, the c.1023insC was a novel mutation and truncated Pendrin protein, which might lead to dysfunction of ion transportation. Our finding is expected to further expand the spectrum of SLC26A4 variation associated with EVA in Chinese population.
MATERIALS AND METHODS
Subjects and clinical evaluations
Two Chinese boys with HL and their normal family members were recruited, including parents and siblings. Particularly in family I, the proband and his normal brother were fraternal twins. Informed consent and blood samples were obtained from all subjects. This study was approved by the Ethics Committees of Beijing Children’s Hospital. The two probands got detailed medical history and physical examination, including thyroid sonography, functional thyroid tests (to exclude Pendred syndrome) and computed tomography (CT) scan of the temporal bone. Clinical audiometric evaluations were conducted, such as play audiometry (PA), auditory steady state response, tympanometry, acoustic spatial reflex, auditory brainstem response and distortion product otoacoustic emission (DPOAE) recording. PA results were applied to estimate the degree of HL and severity level classified as mild (26–40 dB), moderate (41–55 dB), moderately severe (56–70 dB), severe (71–90 dB) and profound (>90 dB) [13]. All CT images in both axial and coronal sections were obtained with 0.625 mm contiguous increments. EVA was diagnosed if the diameter was greater than 1.5 mm at a midway point between the common crus and the external aperture [14].
Analysis of SLC26A4 gene mutation
Genomic DNA from all family members was extracted from 400 μL of peripheral blood samples using the Blood DNA Kit (CWBio, Beijing, China), following the attached protocol. DNA samples and quality were evaluated using a NanoDrop 8000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and 1% agarose gel electrophoresis. Fragments covering all coding exons of SLC26A4 gene were amplified by polymerase chain reaction (PCR) using specific primers (Supplementary Table 1).
The PCR was performed in a total of 20 μL reaction contained 1 μL of genomic DNA (50–100 ng), 10 μL of premix, 0.8 μL of each primer (10 μM) and water by Premix Taq kit (Takara, Tokyo, Japan). Amplification program is as follows: an initial denaturation at 98°C for 30 seconds, 30 cycles of 98°C for 10 seconds (denaturation), annealing at 60°C–66°C for 30 seconds (annealing), 72°C for 20 seconds (extension) and a final extension at 72°C for 10 minutes. The PCR products were subjected to direct Sanger sequencing using an ABI Prism 377 DNA sequencer (Applied Biosystems, Foster City, CA, USA).
Bioinformatic analysis
To compare the sequence homology of SLC26A4 in different species, phylogenetic analysis was performed using MEGA software (Pennsylvania State University, State College, PA, USA). Genetic evolution related species were chosen as previously reported [15], including Homo sapiens (NP_000432.1), Nomascus leucogenys (XP_003268184.1), Pan troglodytes (XP_009 452244.1), Macaca mulatta (XP_001094049.1), Canis lupus familiaris (XP_540382.3), Sus scrofa (XP_003357559.1), Mus musculus (NP_035997.1), Rattus norvegicus (NP_062087.1), Cricetulus griseus (XP_007648133.1) and Xenopus tropicalis (NP_001107135.1).
Deleterious analysis of mutations were performed by online Sorting Intolerant From Tolerant (SIFT) and Polyphen2. Model building and structure analysis were further conducted to predict the functional outcome of the novel variant. Online tool SWISS-MODEL was applied to build 3D modeling of the human SLC26A4 (NP_000432.1). The PyMOL software (DeLano Scientific LLC, San Carlos, CA, USA) was used to visualize the homology modeling and structure analysis.
RESULTS
Clinical features and evaluation
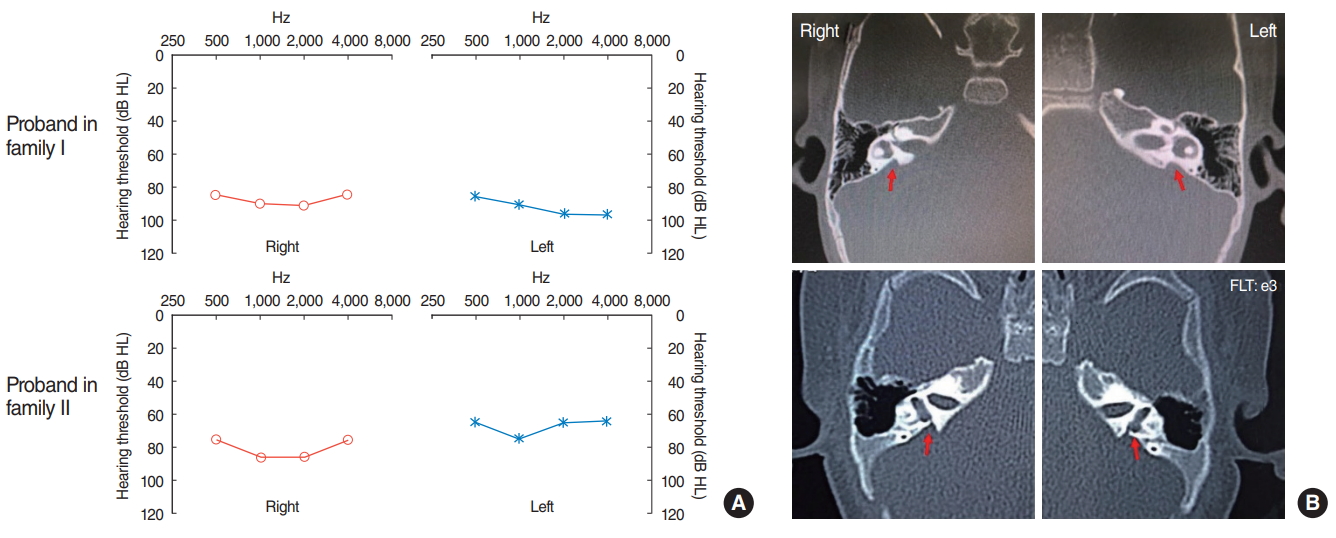
In the present study, two HL subjects from two unrelated families and their normal-hearing family members were enrolled. The first proband (2-year-old boy) was noticed due to significant delays in speech development and diagnosed as HL, while the second proband (4-month-old boy) was identified by newborn hearing screening [7]. As shown in Fig. 1A, both probands suffered from severe sensorineural HL in both ears based on PA detection. Tympanogram was evaluated as “A” and the bilateral acoustic stapedial reflex was not elicited. Auditory brainstem response was not elicited for the first proband, but the thresholds in the second proband were 100 dB HL (right ear) and 70 dB HL (left ear) when motivated at 100 dB HL and 80 dB HL, respectively. In addition, DPOAE reveals no response for both probands in both ears while CT scans of the temporal bone showed EVA bilaterally without cochlea malformations (Fig. 1B). According to normal thyroid function tests, hypothyroidism was excluded. The physical examination and otoscopy were normal. In order to remain hearing ability and avoid speech delay, the first proband had received unilateral cochlear implant and language rehabilitation training. The second proband had received hearing aid and prepared to get cochlear implant. Both probands are in our close follow-up.
SLC26A4 mutation analysis and structure modeling
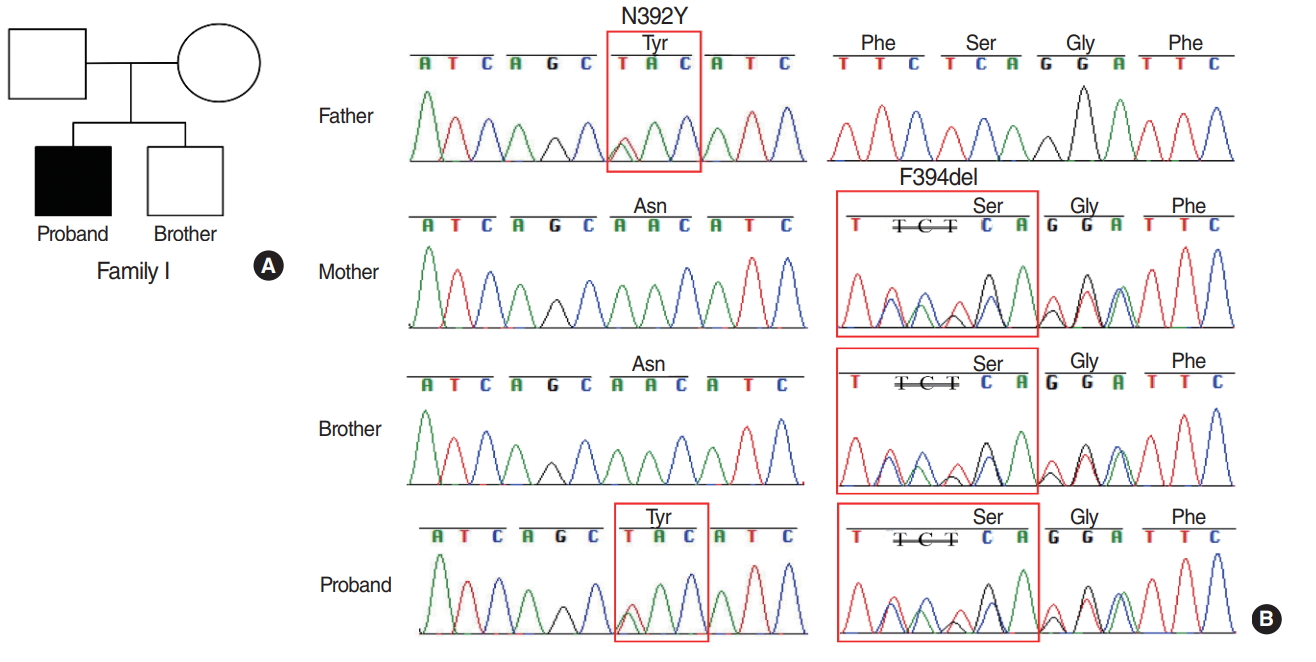
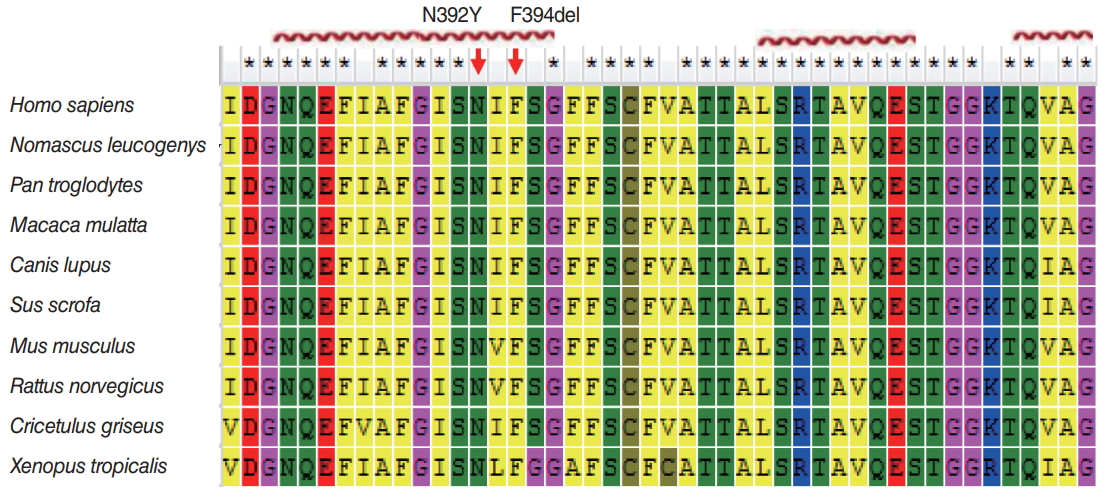
For the proband in family I, sequence analysis of SLC26A4 found a compound heterozygote of c.1174A>T (p.N392Y) missense mutation and c.1181delTCT (p.F394del) in exon 10 (Fig. 2). The father was a heterozygous carrier of 1174A>T mutation, the mother and the elder brother were both heterozygous carriers of 1181 TCT deletion. According to Genome Aggregation Database, both p.N392Y and p.F394del were with low allele frequencies of 4.068e-6. We further analyzed sequence conservation of mutation sites by sequence homology analysis in different species. The results verified that p.N392Y and p.F394del located at the conservative region of the Pendrin protein sequence (Fig. 3). The SIFT analysis predicted p.N392Y to affect protein function (with a score of 0.00) and Polyphen2 showed that p.N392Y was probably damaging (with a score of 1.00). The results strongly suggested that these mutations were closely linked to Pendrin function and thus HL.
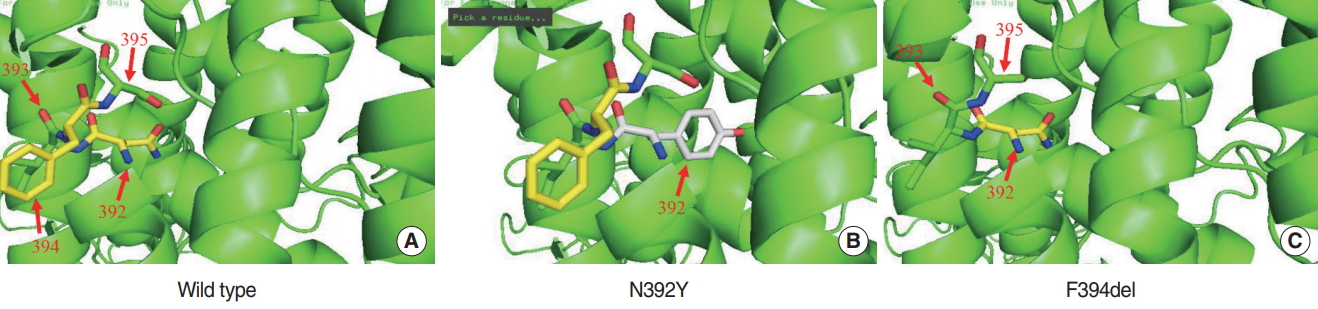
The homology modelling of SLC26A4 (Pendrin) was further constructed by online SWISS-MODEL (PDB ID: 5da0.1.A) [16]. We analyzed the wild and mutant structure (p.N392Y and p.F394del) of SLC26A4 by PyMOL software (Fig. 4). Compared with wild-type structure, p.N392Y mutant protein possess one more benzene ring, while p.F394del mutant protein lacks a benzene ring. Since benzene ring possess relatively large spatial structure, these mutation-induced structure organization change might affect the function of Pendrin protein. Regardless of other genetic changes, these data together with the clinical and genetic presentation, indicated that the co-occurrence of p.N392Y and p.F394del mutations in SLC26A4 might account for the EVA cause of the proband I.
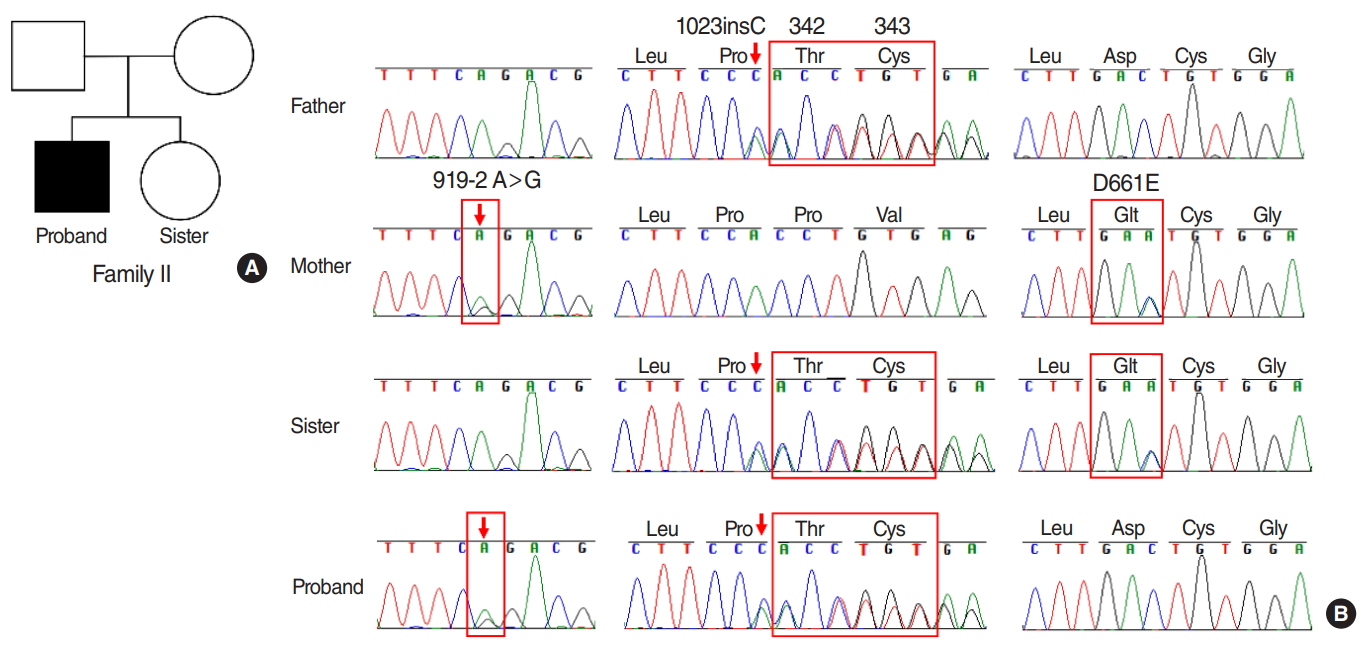
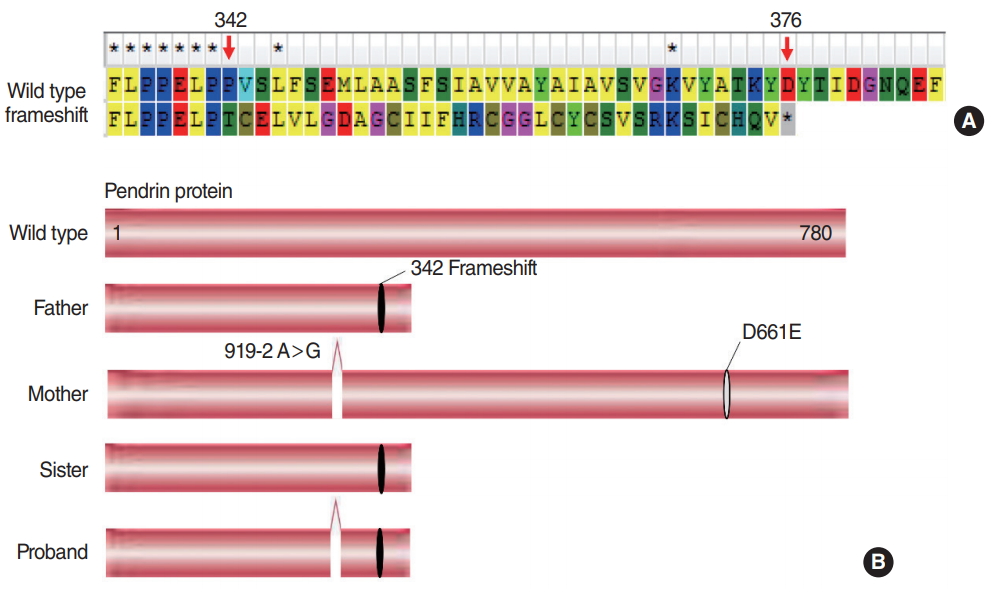
In family II, a compound heterozygote was identified with c.919-2A>G mutation in the splice site of intron 7, a mutation c.1023insC in exon 9 and c.1983C>A (p.D661E) mutation in exon 17 (Fig. 5). The father was a heterozygous carrier of c.1023 insC mutation, the mother was a heterozygous carrier of both c.919-2A>G mutation and c.1983C>A mutation, and the proband’s healthy sister was a heterozygous carrier of c.1023insC and c.1983C>A mutation from her parents, respectively. Both 919-2A>G and D661E were common mutations with relatively high allele frequencies of 3.36e-4 and 9.749e-5. The variant of p.D661E have been reported previously [17], and both SIFT and Polyphen2 analysis showed that p.D661E was deleterious. For c.1023insC, this mutation was considered as the first report and this novel mutation led to a frameshift mutation, caused a stop codon at position of 376 amino acid, resulting in truncated Pendrin protein (Fig. 6A). Functionally, the structure defect might affect its function of ion transportation. The putative schematic representation of Pendrin protein and mutants was shown in Fig. 6B.
DISCUSSION
Two patients were diagnosed as HL from unrelated two families. Both probands were clinically checked inner ear malformation with EVA. SLC26A4 is associated with EVA, to characterize the underlying genetic mechanisms resulting in the presentation, we performed sequence analysis of all coding exons in SLC26A4 on the two patients, their sibling and parents, respectively. We identified two compound heterozygous mutations (c.1174A>T/c.1181delTCT in proband I; c.919-2A>G/c.1023insC in proband П) in Pendrin protein independently. Moreover, SLC26A4 c.1023insC mutation in proband П has not been reported so far, and this novel mutation generated a frameshift and consequent truncated Pendrin protein at position of 376 amino acid, which might contribute to Pendrin dysfunction. Thus this study contributes to improve our understanding of the causes of HL associated with EVA.
Evidences have shown that HL is related to genotype, and biallelic mutations could cause more severe deafness, earlier age of onset, and larger EVA size [12,18]. In Chinese population, GJB2, SLC26A4, mitochondrial 12S rRNA and GJB3 are hotspot genes [19,20]. Mutations in SLC26A4 spread over all its exons, the prevalent mutations differ in different countries and ethnic groups [21-23]. In Han Chinese, SLC26A4 c.919-2A>G and c.2168A>G were the most prevalent pathogenic mutations for HL patients [5,24].
In family I of the present study, we identified mutations of c.1174A>T (p.N392Y) and c.1181delTCT (p.F394del) in SLC26A4 (Fig. 2). To our knowledge, c.1174A>T mutation and c.1181delTCT have been identified before [25,26]; however sequence conservation analysis of these amino acid regions and protein structure analysis are far from clear. Moreover, only one study identified c.1181delTCT (p.F394del) so far, which inaccurately reported p.F394del as p.S394del [26]. In addition, our study was the first time to identify the co-occurrence of c.1174A>T and c.1181delTCT in one patient. Structurally, sequence homology analysis showed that the amino acid sites of both p.N392Y and p.F394del of SLC26A4 are highly conserved in different species (Fig. 3), suggesting that they might be crucial for the Pendrin structure and function.
The SLC26A4 gene encodes the transmembrane protein Pendrin, which acts as a chloride/bicarbonate transporter. Pendrin is associated with the homeostasis of ion concentration in the endolymph of the inner ear. By screening UniProt Knowledgebase, Pendrin p.N392Y and p.F394del are found located at transmembrane site of Pendrin. Previous experimental study have demonstrated that Pendrin p.N392Y mutant localized in the cytoplasm instead of the cellular membrane like wild-type Pendrin, which clarified the consistency between genotype and phenotype [27]. Our further protein structure analysis showed that p.N392Y mutant protein of Pendrin acquired one more benzene ring, while p.F394del mutant protein lacks a benzene ring (Fig. 4). These alterations in protein residues and side chains might result in conformational changes in Pendrin spatial structure, thus cause Pendrin dysfunction, like intracellular localization alteration. In the only study performed in Chinese immigrants to Singapore, the Pendrin p.F394del was also identified [26], suggesting that this deletion mutation might specific in Chinese population. Since of Pendrin p.F394del mutation is also localized on transmembrane site near p.N392Y, it was functionally supposed to be analogous to N392Y mutation in Pendrin and thus anion exchange.
In family II, a novel compound heterozygous mutation (c.919-2A>G/c.1023insC) was found in SLC26A4, and this genotype is consistent with the clinical diagnosis of EVA (Fig. 4). Moreover, the novel mutation of c.1023insC induced a frameshift mutation, and consequently led to truncated Pendrin protein (Fig. 6). This particular variant disrupting important domains such as sulfate transporter and anti-sigma antagonist (STAS) domain. The STAS domain regulates the stability, tracking and anion transport function of Pendrin [28,29]. Well-organized structure has been confirmed critical to maintain the STAS domain. Therefore, it is possible that the novel mutation identified in our study might impair the activity of Pendrin as an anion transporter by protein truncating and conformation altering of the Pendrin protein. As a result, it is clinically beneficial to screen SLC26A4 mutations in patients with HL. Mutations identification in this gene may help us design more cost effective strategies in genetic therapy of HL with EVA. However, the molecular mechanism between this compound heterozygous mutation and the consequent phenotype need further functional studies. Further investigation is necessary to clarify the molecular mechanism between this compound heterozygous mutation and the consequent phenotype.
In summary, we identified two compound heterozygous mutations (c.1174A>T/c.1181delTCT; c.919-2A>G/c.1023insC) in SLC26A4 in two Chinese families with EVA. The novel mutation of c.1023insC in SLC26A4 has not been reported in other countries and regions, which leads to a frameshift mutation, caused a stop codon at position of 376 amino acid, resulting in a truncated deletion of Pendrin protein. Our finding is expected to further expand the spectrum of SLC26A4 mutations associated with EVA in the Chinese population.