INTRODUCTION
Usher syndrome (USH), an autosomal recessive disorder, is the leading cause of hereditary deaf-blindness in humans [1]. USH is a clinically and genetically heterogeneous condition, characterized by bilateral sensorineural hearing loss (SNHL), progressive visual loss due to retinal photoreceptor degeneration presenting as retinitis pigmentosa (RP), and vestibular dysfunction [2]. Based on the severity and progression of hearing loss, the onset age of RP, and the presence of vestibular dysfunction, USH can be classified into three categories [3]. USH type I (USH1) is characterized by severe to profound congenital deafness, vestibular areflexia, and prepubertal onset of RP. USH type II (USH2) is characterized by congenital moderate to severe hearing loss, preserved vestibular function, and RP onset in the second decade of life. USH type III (USH3) presents progressive hearing loss and variable degrees of RP and vestibular response. In addition, some patients could be categorized as suffering from atypical USH if their phenotype is not compatible with one of the three aforementioned classifications [4].
USH2 is the most common type, which is found in more than 50% USH patients [5]. USH2 is known to be associated with three known genes, including USH2A, ADGRV1, and WHRN [6,7]. Recently, PDZD7, which is an encoding PDZ that contains a scaffold protein, was also suggested to act as a modifier and cause digenic inheritance of human USH2 [8]. These four proteins have been shown to constitute USH2 interactome by dynamically interplaying via the PDZ domain [9]. USH2A (OMIM 608400), located on chromosome 1q41, accounts for approximately 80% of USH2 cases. USH2A encodes the usherin, where it demonstrates a wide, but not ubiquitous distribution [10]. Usherin was expressed in various tissues, but primarily localized to the basement membrane of the cochlea and retina, concentrating particularly on the apical inner segment recess that wraps around the connecting cilia in the photoreceptor cells [11,12]. Thus, the alternation of usherin is associated with the postnatal development of cochlear hair cells and retinal photoreceptor cells; USH2A variants have been linked to non-syndromic RP and SNHL, thereby leading to a diverse clinical phenotype [13-15]. Although there have been numerous studies on the functional role of USH2A, the pathophysiological mechanism underlying its phenotypic heterogeneity, to the best of our knowledge, has not been fully elucidated to date. To date, more than 70 different variants of USH2A have been reported in patients of different ethnicities suffering from USH2, including some founder variants [16]. The audiometric configuration caused by USH2A variants typically manifested moderate to severe SNHL, showing a down-sloping configuration characterized by a mild-to-moderate hearing loss at low frequencies and a severe-to-profound hearing loss at high frequencies [17]. However, diverse audiological phenotypes, ranging from mild to severe degree, and a possibility of further progression have been also reported [18]. Although the genotype-phenotype correlation in USH2A variants remains enigmatic, interestingly, a specific combination of USH2A variants causing truncating proteins in both alleles has shown to elicit a more severe and progressive audiological phenotype [19]. Thus, understanding the audiological complexity associated with USH2A may provide insight into the appropriate genetic counseling. We, herein, report two novel USH2A variants associated with severe to profound SNHL mandating cochlear implantation and resultant audiological and ophthalmological phenotypes in two unrelated Korean families. Additionally, we sought to discuss a possible allelic hierarchy.
MATERIALS AND METHODS
Subjects
All procedures in this study were approved by the Institutional Review Boards of Seoul National University Bundang Hospital (No. IRB-B-1007-105-402). Written informed consent was obtained from all participants. Two subjects (SB237-461, male, 46 years and SB354-692, female, 34 years at ascertainment) with suspicion of USH2 visited our clinic for molecular genetic diagnosis. Patients were interviewed via an evaluation of comprehensive clinical history to rule out perinatal, toxic, infectious, and traumatic factors that could cause non-genetic hearing loss or retinal degeneration. We referred all of our subjects to a retinal specialist to examine the progressive visual loss due to RP. Accordingly, the two subjects with genetically confirmed USH2A variants were enrolled in this study.
Audiological evaluation
A comprehensive assessment of SNHL was conducted by the following clinical examinations: audiological tests, including pure-tone audiometry (PTA) and speech evaluation, and imaging tests, such as temporal bone computed tomography (TBCT). PTA, with air and bone conductions at frequencies ranging between 250 and 8,000 Hz, was performed according to the standard protocol. TBCT was employed to identify any inner ear anomalies, such as enlarged vestibular aqueduct (EVA), in these probands. Depending on the PTA results, the hearing loss range was described in the following manner: low frequency, 250–500 Hz; mid frequency, 1–2 kHz; and high frequency, 4–8 kHz. The severity of hearing loss was classified as mild (20–40 dB), moderate (41– 55 dB), moderately severe (56–70 dB), severe (71–90 dB), and profound (>90 dB). The configuration of audiogram was demonstrated to be down-sloping, U-shaped, rising, or flat, in conjunction with the pattern of hearing loss across various frequencies [20]. There was a difference between the left and right ear air condition threshold >20 dB at least two frequencies out of 0.5, 1, and 2 kHz were indicated as asymmetric.
Ophthalmologic evaluation
Two subjects underwent standard ophthalmological examinations, including measurements of best-corrected visual acuity (BCVA), using a decimal visual acuity chart; slit-lamp biomicroscopy; color fundus photography, using a conventional fundus camera or a ultra-widefield fundus camera (Optos PLC; Dunfermline, UK); and spectral-domain optical coherence tomography (OCT; Spectralis, Heidelberg Engineering, Heidelberg, Germany). The full-field electroretinogram (ERG; VERIS, Electro-Diagnostic Imaging Inc., Redwood City, CA, USA) was performed according to the six standard protocols recommended by the International Society for Clinical Electrophysiology of Vision. Goldmann perimetry was performed under the isopter of the III/4e white test light. The ERG and Goldmann perimetry were performed in patient SB237-461.
DNA preparation and molecular genetic testing
Whole-exome sequencing (WES) from two subjects and a bioinformatics analysis of the data were employed, as previously described [21,22]. The obtained reads were aligned with the UCSC hg19 reference genome (https://genome.ucsc.edu/), which is an interactive website with the genome sequence data of a variety of vertebrate and invertebrate species, including major model organisms, integrated with a large collection of aligned annotations, with narrowed down variants. Bioinformatics analysis was conducted to select the potential candidate variants. In brief, the analysis was conducted in the following manner. the raw data generated by WES was mapped onto the UCSC hg19 reference genome and rare single nucleotide variations (SNVs), indels, or splice site variants were selected, via the following filtering process: (1) a basic filtering step, which entailed an exclusion of synonymous SNVs, and a selection for SNVs with a quality score of greater than 30 and a read depth of more than 20; (2) compatibility with the AR inheritance pattern; (3) confirmation of the presence of variants by Sanger sequencing; (4) a segregation study and/or control study against the Korean Reference Genome Database (KRGDB) consisting of 1,722 Korean individuals (3,444 alleles) (http://coda.nih.go.kr/coda/KRGDB/index.jsp); and (5) compatibility with clinical features. SNVs with the minor allele frequency (MAF) ≤0.005 were included in the study. The global MAF was checked using several databases, including 1,000 Genomes, Exome Aggregation Consortium (ExAC). To analyze the possible pathogenic potential of the detected variants, according to inheritance pattern and audiogram patterns, SIFT (http://sift.jcvi.org/), PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), and/or Mutation taster (http://www.mutationtaster.org/) were used. Additionally, to estimate the evolutionary conservation of the amino acid sequences, we referred to the GERP++ score from the UCSC genome browser (http://genome.ucsc.edu/).
RESULTS
Audiological and vestibular manifestations
We identified two unrelated Korean probands, presenting both more than severe SNHL with down-sloping configuration and a hint of suspicious RP. The inheritance pattern was presumed to be autosomal recessive or sporadic. A 46-year-old male (SB 237-461), who was regularly followed-up for RP examination, was referred to our clinic due to bilateral hearing impairment since childhood. He used hearing aids since he was in his 20’s, but mainly relied on lip-reading for communication. Based on PTA, severe to profound SNHL was documented with down-sloping configuration (Fig. 1A).
Additionally, a 34-year-old female (SB 354-692) suffering from bilateral hearing difficulties since childhood and night blindness visited our clinic. Hearing deterioration had been aggravated since she was in her 20’s and has been wearing hearing aids since then. Based on PTA, profound SNHL on the right ear and severe to profound SNHL on the left ear were documented with down-sloping configuration (Fig. 1B).
In this study, the two affected patients were planning to undergo cochlear implantation due to insufficient speech performance even after wearing hearing aids. Moreover, between the two unrelated probands, there were no inner ear anomalies, such as EVA, which may precipitate in more severe and progressive hearing loss in TBCT.
None of the two probands had subjective dizziness. The comprehensive vestibular examinations were available only in SB237-461 (Supplementary Fig. 1). During an exhaustive videonystagmography examination (SMI, Teltow, Germany or Ics Medical, Schaumburg, IL, USA), no specific nystagmus was noted on spontaneous nystagmus on both with and without fixation, gazeevoked nystagmus, head-shaking nystagmus, and vibration-induced nystagmus test. The bithermal caloric test (Ics Medical) result showed no caloric paresis. Cervical vestibular-evoked myogenic potentials (NI PCI-4461; National Instruments, Austin, TX, USA) test showed normal symmetric response. Moreover, neither overt nor covert saccadic movements were found in video head impulse test (SLMED, Seoul, Korea), and the vestibulo-ocular reflex gains of both lateral semicircular canals were intact.
Ophthalmologic manifestations
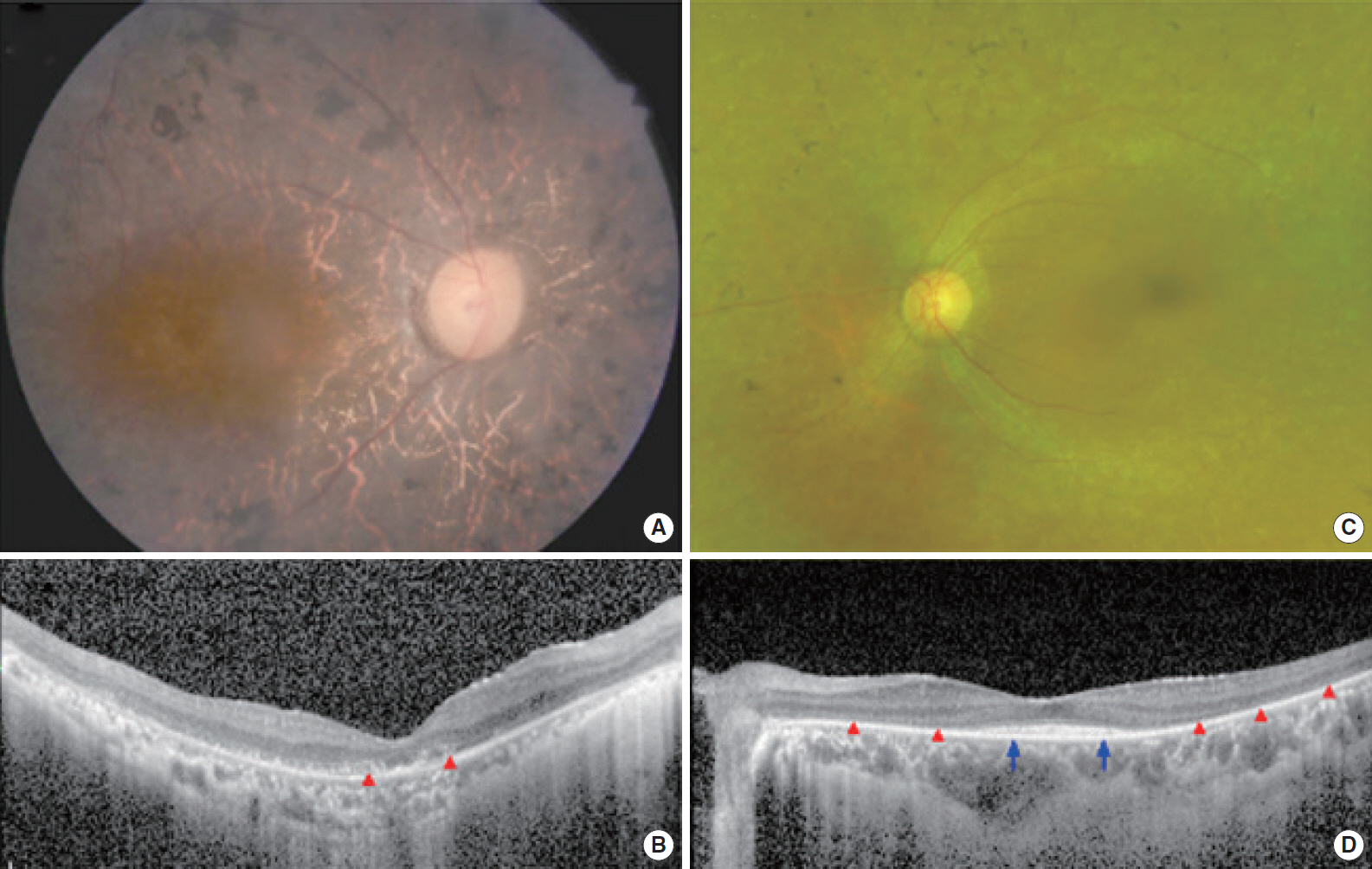
SB237-461 has being suffering from night blindness since the mid-teens, and his visual acuity has been decreasing gradually since he was about 30 years of age. After the recent cataract surgery, BCVA was 20/125 in the right eye and 20/100 in the left eye (Table 1). Fundus examination showed typical features of RP, including diffuse pigmentary changes of the retina, vascular attenuation, and waxy disc pallor in both eyes (Fig. 2A). Diffuse disruption of retinal photoreceptors, including the macula, was observed on the OCT examination (Fig. 2B). In accordance with anatomical abnormalities, the ERG response was extinguished in all records, and the overall loss of visual field was observed except for a small central island within 5° of the central visual field. SB354-692 mainly complained of night blindness. Her BCVA was 20/32 in the right eye and 20/30 in the left eye. Diffuse degenerative changes were observed in the peripheral retina (Fig. 2C). Compared with the disruption of the photoreceptors in the pericentral area, central photoreceptors were relatively preserved on the OCT examination (Fig. 2D). The clinical features of these two patients were consistent with the typical manifestations of RP without any past history of other suspected retinal disorders.
Molecular genetic diagnosis
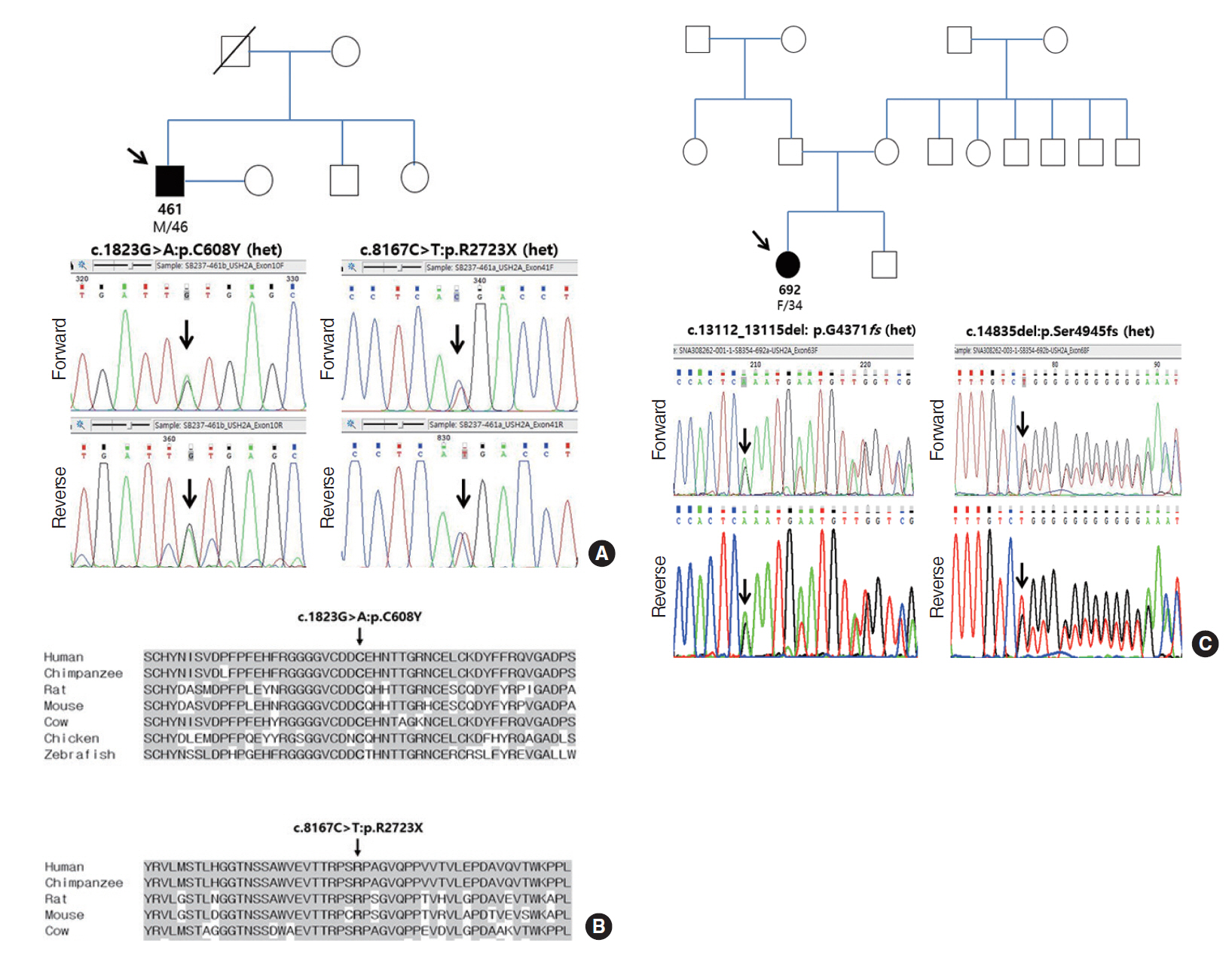
After filtering the potential candidate variants, four autosomal recessively inherited variants of the USH2A gene were determined to possess pathogenic potential. The variants were confirmed via Sanger sequencing chromatograms. Two (c.1823G> A: p.C608Y, c.14835delT: p.Ser4945fs) of the variants were found to be novel pathogenic variants (Table 2).
In SB 237-461, two possible candidate genes, including USH2A and STRC, were initially screened. A nonsense variant (single heterozygote) of the STRC gene, p.Q195X, was detected as a single heterozygote and was predicted to be non-pathogenic. Multiplex ligation dependent probe amplification assay (MLPA) was performed using the SALSA MLPA P461 DIS probemix kit (MRC Holland, Amsterdam, Holland) for STRC. In addition to STRC variants, large deletion in the STRC gene was screened using MLPA. There was no copy number variation in STRC exon 19 to 25 (Supplementary Fig. 2). Consequently, two variants of USH2A were determined to be pathogenic variants (Fig. 3A). A novel missense variant, c.1823G>A: p.C608Y, of the USH2A gene, was predicted to be pathogenic, as “damaging” or “probably damaging,” via SIFT and polyphen-2 analyses. Furthermore, the residue of p.C608Y was evolutionarily well-conserved among several species and the pathogenic potential of this variant was also supported by the high GERP++ score of 4.61 and phyloP score of 4.66 (Fig. 3B). In addition, a previously reported nonsense variant (p.R2723X) was also predicted to be pathogenic because prediction software (mutation taster) was used to predict the variant that may affect the protein function by showing the result as “disease causing” (with a score of 1) [23]. For p.R2723X, residues with GERP++ score of –0.488 and a phyloP score of 0.953 were defined as non-conserved but exhibited a global MAF value of 0.00002 (via 1,000 G). In SB 237-461, the two variants were not detected based on the ethnically matched normal population and KRGDB. According to ACMG/AMP standards and guidelines [25,26], p.C608Y and p.R2723X, meets the criteria for “uncertain significance” and “pathogenic” respectively. As for a novel missense variant (p.C608Y), this call is supported by one moderate evidence and two supporting evidence (Table 2). This variant was rare enough to satisfy rarity in population databases, leading to PM2. Additionally, PP4 with a highly specific patient’s phenotype and PP3 (REVEL score of 0.918) could be applied. Resultantly, p.C608Y can be classified as “uncertain significance” according to the combining criteria to classify variants [27] For a previously reported truncating variant (p.R2723X), this call is supported by one very strong evidence (PVS1), two moderate evidence (PM2 and PM4), and one supporting evidence (PP4) (Table 2).
In SB 354-692, based on WES, the LOXHD1 and USH2A genes were screened as possible candidate genes. Among the three candidate variants, two frameshift variants in USH2A, p.Ser4945fs, and p.Gln4371fs, were predicted to be pathogenic variants (Fig. 3C). A novel variant, p.Ser4945fs, was not detected based on KRGDB and global MAF and was evolutionarily well-conserved among several species by the high GERP++ score of 5.19. The previously reported truncating variant, p.Gln4371fs, exhibited a KRGDB value of 0.00029 [27]. According to ACMG/AMP standards and guidelines, these two variants meets the criteria for “pathogenic,” which is supported by one very strong evidence (PVS1), two moderate evidence (PM2 and PM4), and one supporting evidence (PP4) (Table 2).
DISCUSSION
Here, we reported the two novel variants of USH2A, including one missense variant (c.1823G>A: p.C608Y) and one frameshift variant (c.14835delT: p.Ser4945fs), in two unrelated Korean families segregating SNHL and RP in autosomal recessive fashion. According to ACMG/AMP standards and guidelines, two novel variants in USH2A, p.C608Y and p.Ser4945fs, meet the criteria for “uncertain significance” and “pathogenic,” respectively. Interestingly, the resultant audiometric phenotype showed a severe or more than severe SNHL with down-sloping configuration necessitating cochlear implantation.
The variants in USH2A have shown to exert a broad spectrum of clinical heterogeneity [13]. For example, in five Chinese families, patients carrying the compound heterozygous variant (c.2299delG and p.C759F) showed atypical USH, whereas those carrying homozygous c.2299delG and p.C759F variant exhibited typical USH2 and non-syndromic RP, respectively [28]. In addition, the phenotypic variations were reported even in the same variant of USH2A (c.2314delG) [29]. Clinical reports have also provided another excellent example of this allelic hierarchy, where two CDH23 alleles with truncation variants led to USH type 1D, while one CDH23 truncated allele and one CDH23 missense allele led to DFNB 12, a nonsyndromic type of recessively inherited SNHL [30].
Diverse audiological phenotypes from mild to profound hearing loss have been reported in USH2A variants [18,19,31], although most patients exhibited a moderate degree of hearing loss. By reporting two novel variants of USH2A, our findings provide novel insights regarding the expansion of audiological spectrum from moderate to severe SNHL to more than severe SNHL in Koreans, supporting the diverse auditory phenotype of USH2. Theoretically, given a wide distribution of variants along the USH2A domains and relatively narrow audiological dynamic range in USH2 compared with that in USH1, the genotype-phenotype correlation of USH2A has not been fully established. However, it has recently been suggested that there is evidence of genotype-phenotype correlation for some known USH2A variants. In detail, based on a large cohort study evaluating patients diagnosed with USH, most carriers of p.Glu767Serfs*21 variant showed moderate deafness, whereas patients with p. Cys759Phe variant exhibited only mild hearing loss [31]. Furthermore, a recent study suggested that an alternative allele may contribute to the variation in auditory manifestation [18]. Based on the four Swedish families, the same heterozygous variant (c.2299delG) in one allele showed an inter- or intra-familial difference in terms of the degree of hearing loss [18]. In line with this, p.R2723X in SB237-461, a previously identified nonsense variant associated with non-syndromic RP or typical USH2 in patients from North America [15], was found to have severe to profound SNHL, supporting the differential phenotypes based on their genotype combination.
Previous studies which investigated audiological findings in USH2 patients demonstrated a lack of genotype-phenotype correlation [32,33]. Recently, a combination of two truncating variants, including homozygotes for the c.2299delG mutation, is likely to develop more severe and progressive hearing loss in subjects carrying the USH2A variants [19]. Interestingly, this seems to be consistent with our results: the SB354-692 carrying two truncating variants demonstrated more severe hearing threshold than the SB237-461 carrying one truncating mutation and one missense mutation, suggesting a potential genotype-phenotype correlation Considering that most patients with USH2A exhibited a milder progression of hearing impairment ranging from annual 0.4 dB at the lower frequency level to annual 0.8 dB at the higher frequency level on the basis of a cross-sectional linear regression analysis [19], the hearing threshold across the all frequencies of a relatively young SB237-461 (F/32) can be considered as worse than SB237-461 (M/46). Indeed, the truncating variants lead to the loss of a large part of the protein molecule due to premature termination of USH2A translation or deletion of more than one amino acid residue, whereas a missense substitution results in a significant change of polarity at the highly conserved region [34]. We, previously, have raised the hypothesis that the auditory phenotypic severity of DFNB12 could be determined based on genotypic hierarchy of CDH23, implying allelic hierarchy [35]. That is, those with compound heterozygosity of a known pathogenic DFNB12 allele in trans configuration with CDH23 allele with unknown pathogenicity might be related to more severe auditory phenotype than those with the compound heterozygosity of two CDH23 alleles with unknown pathogenicity [35]. Although allelic hierarchy seems to not necessarily the limited notion used to distinguish between nonsyndromic and syndromic phenotype, the term of allelic hierarchy in terms of auditory phenotypic variability depending on the genotypic hierarchy awaits further confirmation, due to lack of supporting evidence.
However, this potential genotype-phenotype correlation in USH2A may be applicable more to hearing loss than to RP. In contrast to the audiological phenotype, SB237-461 carrying one truncating mutation and one missense mutation appears to manifest a more severe visual acuity based on BCVA than SB354-692 carrying two truncating variants. This discrepancy might be explained by different age at RP diagnosis in two subjects. Most patients with USH2A-associated RP showed a relatively preserved visual acuity in their thirties or forties, but a higher cumulative risk of visual impairment in their fifties [36,37]. A recent study suggested that subjects with truncating variants could have earlier onset or rapid deterioration of visual phenotype than those with missense mutations [38]; however, this finding was limited by a comparison of the visual phenotypes between subjects carrying truncating variants and subjects with only missense variants. It may be possible that the visual phenotype converges to the phenotype of the worst variant, while the hearing phenotype can reflect the sum of all phenotypes of the two mutant alleles. Based on this, further investigation, including longitudinal follow-up, is necessary to determine whether the potential genotype-audiological phenotype was replicated in the visual phenotype.
Although it is known that SNHL in USH2 subjects remains relatively unchanged, most patients with USH2A exhibited a milder progression of hearing impairment, as shown by previous literature [17]. Moreover, approximately 7% subjects developed severe progression of hearing loss over time and underwent cochlear implantation due to insufficient speech intelligibility with hearing aids [39]. Cochlear implantation has been implicated for better speech intelligibility and quality of life in some patients with USH2A [39]. Given this, such information regarding the possibility of progression to more than severe SNHL should be thoroughly considered for newly diagnosed subjects with USH2A variants. However, further investigation is necessary because genetic modifiers, epigenetics, and environmental factors hinder the genuine association between genotype and phenotype in USH2A.
The present study has some limitations. First, parental segregation study was not available due to a flat refusal. Thus, we could not exclude the possibility whether compound heterozygosity in each proband is in cis configuration (inherited from the same parent) or de novo variants. Although we could not perform the segregation study, it is possible to argue that the variants in each proband are causative to be USH2 by the following logics. The manifestations of two probands in our study are compatible with USH2, characterized by a definite RP, SNHL since childhood, and absence of vestibulopathy. Moreover, the pathogenicity of the USH2A variants was determined as “uncertain significance” or “pathogenic” based on ACMG/AMP standards and guidelines and in-silico prediction analysis. Although the classification for p.C608Y is close to uncertain significance according to ACMG/AMP guideline, this variant may have highly pathogenic potential on basis of several circumstantial evidences. Furthermore, neither hearing loss nor eye symptoms presented in their parents, there might be a paucity of evidence of in cis configuration. That is, the inheritance pattern was presumed to be autosomal recessive or sporadic in the two unrelated families. Also, a recent study reported that de novo mutations were present in at least three out of 28 isolated RP cases, suggesting de novo dominant mutations appear to play a significant role in patients with isolated RP [40]. However, to the best our knowledge, de novo dominant mutations were not found in affected subjects with typical USH2 phenotype. Second, followup hearing data of our probands is absent. At present, suggesting the genotype-phenotype correlation based on our cross-sectional results seems to be an exaggerated interpretation. Therefore, a longitudinal follow-up study is warranted to clarify the genotype-phenotype correlation in subjects with USH2A.
The present study reported two novel variants of USH2A associated with severe or more than severe SNHL in Korea, revealing some insight into the extended audiological spectrum. A specific combination of USH2A variants causing truncating proteins in both alleles could demonstrate more severe audiological phenotype than that of USH2A variants carrying one truncating mutation and one missense mutation, suggesting a possible genotype-phenotype correlation. Based on our findings, severe or more than severe SNHL should be taken into consideration clinically in subjects carrying USH2A variants.
HIGHLIGHTS
▪ We report two novel USH2A variants from two probands manifesting more than severe sensorineural hearing loss (SNHL) and retinal problems.
▪ USH2A should also be included in the candidate gene list of cochlear implantees.
▪ A specific combination of USH2A alleles may contribute to the differential expressivity of SNHL, suggesting a possible genotype-phenotype correlation.
▪ The understanding of audiological complexity associated with USH2A will impact clinical practice in a way that would change genetic counseling and treatment strategy.