INTRODUCTION
Juvenile hyaline fibromatosis (JHF) is an extremely rare disease and less than 70 cases of JHF have been reported worldwide (1). The current name, JHF, was proposed by Kitano in 1976 (2). JHF is an autosomal recessive disease that is usually detected in infancy or early childhood. Most JHF patients exhibit gingival hypertrophy, contracture of various joints, abnormal development of the extremities, osteolytic lesions and pinkish maculopapular lesions of the skin. These skin lesions may involve the nose, cheek, ears, scalp, back or knees; they are not painful, but they can result in cosmetic sequelae (3, 4).
Impaired collagen synthesis is thought to be the major pathogenetic mechanism that's responsible for this disease. Two separate forms of the disease have been proposed: a localized form that's characterized by very slow growth of lesions and a diffuse form that's characterized by large and rapidly growing tumors (5). The case presented here was the diffuse type and this is the first reported case of the diffuse type of JHF in Korea. This study was conducted with the approval of our institutional review board.
CASE REPORT
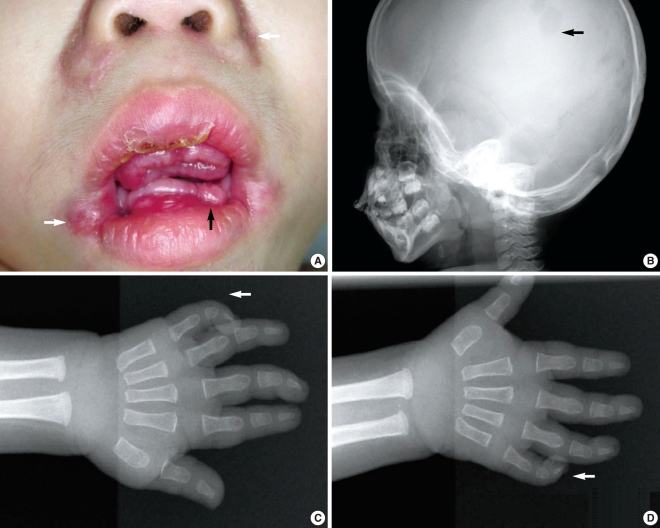
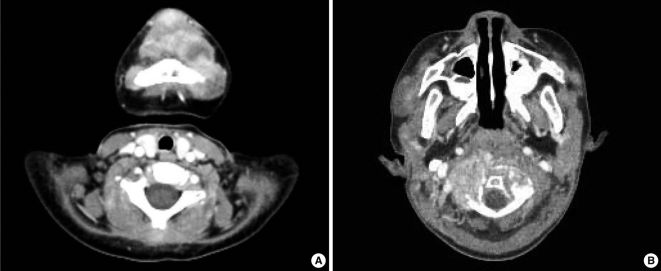
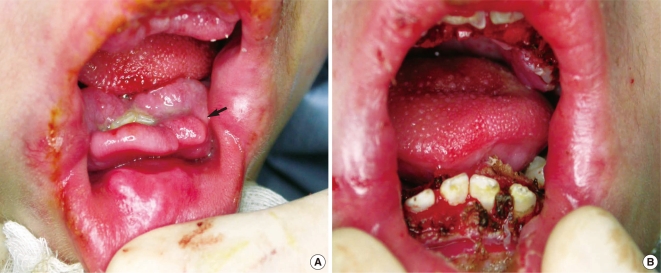
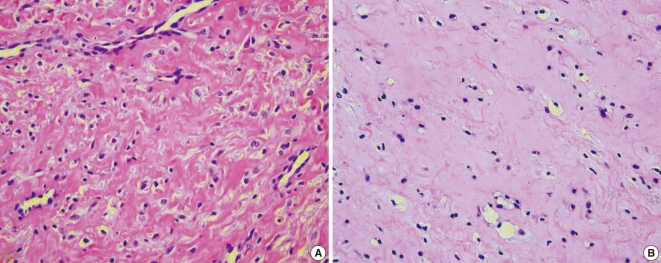
A Korean boy was delivered by cesarean section following an uneventful, full-term pregnancy. There was no familial history of consanguineous marriage. He weighed 2,350 g at birth and he was treated in the neonatal intensive care unit for respiratory difficulty. There was no other abnormality at birth. At 3 months of age, dislocation and stiffness of the hip was noted, and he was treated with an orthosis for 6 months along with additional rehabilitation therapy because of multiple joint contractures. Gingival hypertrophy had developed at 10 months and this caused difficulties with oral feeding. At the time of presentation, the first skin lesion had appeared in the perianal area. Because the perianal mass was initially thought to be a hemorrhoid or an abscess, the patient was treated with sitz bath therapy. At 20 months, he visited the dentist for the treatment of his gingival hypertrophy and he was finally referred to our institution for the removal of a soft, painless, slow-growing mandibular mass that had developed at 12 months and it had grown continually for the past 6 months. The physical examination revealed a soft mandibular mass extending from the lower lip to the mandibular tip. A pinkish maculopapular skin lesion was observed in the submandibular area and the anterior neck, and a soft, 1-cm nodular lesion was observed at both nasolabial folds. In addition, severe gingival hypertrophy and labial nodules were observed in the oral cavity (Fig. 1A). Simple radiography showed multiple osteolytic lesions in the skull (Fig. 1B). The fourth and fifth fingers, both knees and the pelvic joints showed flexion contractures (Fig. 1C and 1D). An enhancing nodular lesion was observed in the submental area via computed tomography (Fig. 2A). Excisional biopsy of the submandibular mass and a gingival hypertrophic lesion was performed under general anesthesia when the boy was 22 months old (Fig. 3). Upon periodic acid-Schiff (PAS) staining and microscopic examination, elastic tissue was rarely observed within the PAS-positive matrix. The fibroblasts were widely distributed and embedded in an abundant amorphous eosinophilic matrix. The PAS-stained hyaline material appeared pink (Fig. 4A). Some stromal cells were observed in the lacuna-like spaces within this material (Fig. 4B). The final pathological diagnosis was reported as JHF. During the follow-up, the gingival hypertrophic lesions and the submandibular masses became larger and they also increased in number. New scalp masses had developed as well. The submandibular mass eventually destroyed the mandible and it invaded the C1 vertebra (Fig. 2B). The gingival hypertrophy impeded oral feeding. When the boy was 32 months old, surgical management that included mass excision and gingivectomy was attempted again. The histological examination showed that these lesions were consistent with the diagnosis of JHF. Despite intermittent hospitalization, the patient's general condition gradually deteriorated because of nutritional deficiency, ascites, recurrent pneumonia and anemia. Recurrent pneumonia and muscular degenerative changes aggravated the boy's respiratory failure. Sadly, the patient eventually died of hypoglycemia and cardiogenic shock at 35 months of age while on supportive care.
DISCUSSION
JHF is clinically characterized by pinkish papules or subcutaneous nodules, osteolytic lesions and gingival hypertrophy. Gingival hypertrophy is a common finding and as demonstrated here, this can be severe enough to interfere with proper feeding and eventually mastication, which can in turn lead to malnutrition, recurrent infections and even death (6, 7). Flexion contracture involving the knees, elbows, fingers and hip joints may occur when this type of tumor infiltrates the joint capsule.
The primary treatment for the skin and oral lesions is surgical excision. Radiotherapy was not effective and the long-term risks of using this modality for treating young patients are well understood. Symptomatic management of the oral lesions includes the extraction of the mobile and carious teeth, and performing gingivectomy provides only short-term benefit and this typically requires several treatments (6). Joint contracture is often associated with a poor prognosis, and capsulotomy, physiotherapy and corticosteroid therapy have all been applied with only modest success (2, 7, 8). The improvement via capsulotomy is known to be temporary, and the patient can be left with permanent joint contracture and deformity (2).
The laboratory findings for JHF are nonspecific and so they of little diagnostic use. Hypochromic anemia has been reported in several of these patients and it is likely related to malnutrition (9). The differential diagnosis inclu-des gingival fibromatosis, infantile systemic hyalinosis (ISH), congenital generalized fibromatosis and Winchester syndrome. Gingival fibromatosis is limited to the gingiva and the lesions typically consist of connective tissue that is rich in collagen I (10). ISH has very similar clinical manifestations and histological findings. However, ISH also includes diffuse thickening of the skin, the development of hyperpigmented plaques on bony prominences, diarrhea as a result of hyalinized growths in the gastrointestinal tract, frequent severe infections and a fatal outcome (11). Congenital generalized fibromatosis is characterized by multiple dermal and subcutaneous nodules, visceral involvement and death shortly after birth (12). Finally, Winchester syndrome is characterized by a short stature, generalized osteoporosis, contractures of small joints and corneal opacities. Skin lesions are uncommon and there is no deposition of a hyaline matrix (3). For our patient, the diagnosis of JHF was based on the presence of typical tumoral skin lesions, gingival hyperplasia, osteolytic bone lesions, joint contracture, muscle weakness and the characteristic histopathological findings. The presence of amorphous hyaline material in the dermis supported our diagnosis of JHF (13-15).
Among the reports concerned with JHF, a few of them described that the patients died of their disease. Our patient had been seen by several departments in our hospital since the age of 3 months old, but the final diagnosis was eventualry made at 22 months old by otolaryngologists. At that time, the disease had already infiltrated so many organs upon presentation, including the scalp, fingers, face and bones, and we had difficulty in providing approprlate management. Intralegional steroid injection wasn't considered because JHF patients showed a limited response in the early disease stage (2). Gingivectomy temporarily improved the amount of oral intake, but was not enough to recover patient's nutritional deficiency. When the malnutrition was not improved sufficiently via gingivectomy, parenteral feeding such as tube feeding, gastrostomy and placing a central line should be considered. The chance to achieve a good result from surgery is reduced as the disease becomes more advanced. Our case proved that Early surgical excision that's performed when the lesions are small has proved most effective. Therefore, making an early diagnosis and administering early treatment are important and we believed that otolaryngologists have a key role to make a diagnosis and manage these patients' oral feeding.
Some studies have reported strong evidence for the presence of a deleterious mutation in the capillary morphogenesis gene 2 (CMG2) located on chromosome 4q21 (16, 17). Identifying the gene mutation that is responsible for this condition will be important in the future as this will give researchers insight into the fundamental defect that is responsible for this hard-to-treat malady. This may be the key to providing physicians with advanced and effective treatment for helping these patients.
JHF is a very rare genetic disease. Physicians should consider the diagnosis of JHF when a young patient presents with multiple maculopapular skin lesions, gingival hypertrophy and joint contracture. Early diagnosis and surgical intervention, including the proper supportive care, may be the best strategy for managing this rare disease.