The Role of Inflammatory Mediators in the Pathogenesis of Otitis Media and Sequelae
Article information
Abstract
This review deals with the characteristics of various inflammatory mediators identified in the middle ear during otitis media and in cholesteatoma. The role of each inflammatory mediator in the pathogenesis of otitis media and cholesteatoma has been discussed. Further, the relation of each inflammatory mediator to the pathophysiology of the middle and inner ear along with its mechanisms of pathological change has been described. The mechanisms of hearing loss including sensorineural hearing loss (SNHL) as a sequela of otitis media are also discussed. The passage of inflammatory mediators through the round window membrane into the scala tympani is indicated. In an experimental animal model, an application of cytokines and lipopolysaccharide (LPS), a bacterial toxin, on the round window membrane induced sensorineural hearing loss as identified through auditory brainstem response threshold shifts. An increase in permeability of the blood-labyrinth barrier (BLB) was observed following application of these inflammatory mediators and LPS. The leakage of the blood components into the lateral wall of the cochlea through an increase in BLB permeability appears to be related to the sensorineural hearing loss by hindering K+ recycling through the lateral wall disrupting the ion homeostasis of the endolymph. Further studies on the roles of various inflammatory mediators and bacterial toxins in inducing the sensorineumral hearing loss in otitis media should be pursued.
INTRODUCTION
The immune system responds to injury or irritation through an innate cascade known as inflammation. There are many cellular and biochemical events which occur as a result of tissue injury which are responsible for the outcome of inflammation. Central to the formation of inflammation are the inflammatory mediators, which include proteins, peptides, glycoproteins, cytokines, arachidonic acid metabolites (prostaglandins and leukotrienes), nitric oxide, and oxygen free radicals. These compounds are produced by epithelial cells, endothelial cells, and infiltrating inflammatory cells. Inflammatory mediators are a double-edged sword, having the potential to fight off infection, but also to damage the host.
Otitis media (OM) is an inflammatory response to either acute or persistent stimuli, typified by the accumulation of both cellular and chemical mediators in the middle ear cleft (1). These mediators have a role in the inflammatory processes of the middle ear such as vascular permeability changes, chemotaxis, stimulation of epithelial secretory activity, enhancement of mucous glycoprotein secretion, and production of other mediators. This paper aims to review the important studies of inflammatory mediators in the pathogenesis of OM and cholesteatoma in order to elucidate which mediators are the most significant. In addition, this paper reviews studies that have identified possible key factors responsible for the chronicity of OM.
INFLAMMATION AND OM
Initiation of inflammation
OM is frequently caused by bacteria which enter the middle ear from the nasopharynx via the eustachian tube. In cases of bacterial otitis media, the source of bacteria is currently believed to be the nasopharyngeal tonsils (adenoids). However, cases of sterile OM are not uncommon and the mechanism of this condition is currently unclear. Endotoxin, a component of bacterial cell walls, is believed to be responsible for initiating inflammation in the middle ear. Endotoxin is a potent inducer of various inflammatory mediators, as well as a modulator of the immune response, and stimulates local macrophages to produce tumor necrosis factor-α (TNF-α) and interleukin-1β(IL-1β). In addition, keratinocytes have the ability to produce many soluble mediators independently from immune cells in response to injury, including TNF-α, IL-β, IL-1, IL-6, and IL-8 (2-5). Many of these cytokines contribute to the acute phase of the inflammatory response, prime the immune system for rapid activity, and promote the release of other cytokines (Fig. 1).
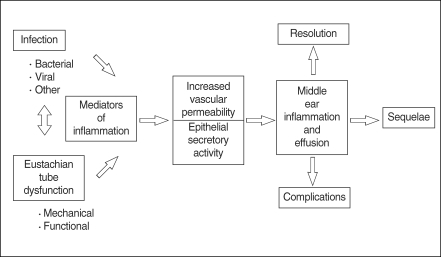
Pathogenesis of otitis media.
Sources of inflammatory mediators
Inflammatory mediators important in OM are produced by infiltrating immune cells such as neutrophils, monocytes, and lymphocytes. In addition, local cells such as keratinocytes and mast cells have been shown to produce inflammatory mediators. It is also coming to light that besides the middle ear, the inner ear tissues are able to produce inflammatory cytokines and use NF-κB activation (6). The sources and functions of inflammatory mediators involved in OM are summarized in Table 1.
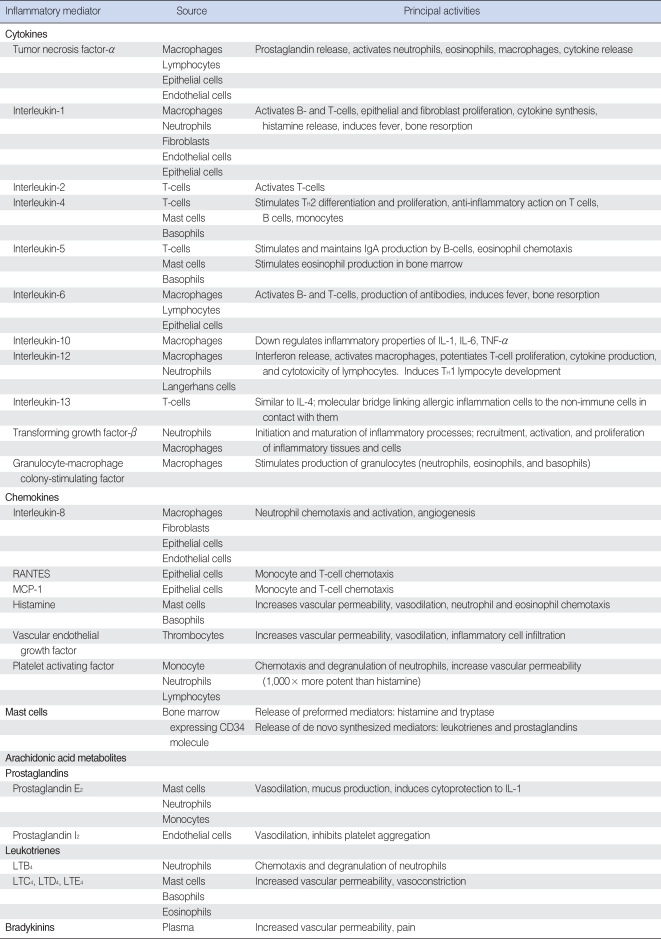
Sources and functions of inflammatory mediators important in otitis media
Complements
Complement is one of the first mediators activated in the initiation of inflammation throughout the body and in OM. This pathway can act as the first cytotoxic response to invasion while also initiating recruitment of leukocytes and vascular changes. Complement is part of the humoral immune system and consists of multiple components that when activated, usually through cleavage, interact to form enzymes or function as binding proteins. Intravascular activation of complement is initiated via a variety of agents including antigen-antibody complexes, bacterial products, and toxins; injured endothelial cells can also activate the complement system (7). The complement system is organized as two pathways with C3 occupying the central position in both. Binding of antibody to antigen or to foreign molecules results in activation of the antibody-dependent classical pathway, whereas the alternative pathway is activated by the lipopolysaccharide (LPS) portion of endotoxin and other nonprotein agents. C3 is biologically inactive, but cleavage by C3 convertase yields active fragments including C3a and C3b. Deposition of the active cleavage product C3b on the surface of foreign particles or target cells allows recognition by receptors on phagocytic cells. C3a is a potent activator of mast cells and basophils and leads to release of histamine from secretory granules. Proteolytic cleavage of C5 occurs via both pathways and generates C5a, a potent mediator of inflammation. C5a is the major complement-derived chemotactic agent for neutrophils, eosinophils, monocytes, and macrophages. C5a also has the ability to activate platelets, leading to their aggregation and surface expression of P-selectin.
Complement activation has been observed in human middle ear effusion by some authors (8, 9). In animal models of acute otitis media (AOM), animals were treated with cobra venom to deplete complement and then their middle ears were antigenically challenged (10). Complement-depleted animals had significantly less inflammation and decreased volume of effusion when compared to controls. Middle ear mucosa from children with chronic otitis media (COM) with effusion has shown large amounts of complement fragments C3 and C9 when analyzed by immunofluorescence microscopy (11). In humans, deficiencies of the complement system are known to cause recurrent or COM (12). Despite this fact, it appears that complement itself may be harmful to the middle ear mucosa. Membrane cofactor protein (MCP) and protectin (CD59) are two middle ear proteins which help prevent unrestricted complement damage (11).
Cytokines
Cytokines are glycoproteins, produced by inflammatory cells and epithelial cells, which modulate the immune response. Cytokines extensively conduct inter-cell communication. Inflammatory cells including neutrophils, macrophages, and lymphocytes use cytokines to coordinate all stages of the inflammatory response. Production of cytokines is conducted by a wide variety of cell types. For example, IL-1 is produced and secreted by macrophages, lymphocytes, vascular endothelial cells, neutrophils, fibroblasts, and monocytes. There are many biological effects including: chemotaxis of cytotoxic T-cells and B-lymphocytes, cytokine synthesis of IL-2 and IL-8 and TNF, neutrophil chemotaxis and degranulation, fibroblast and epithelial proliferation, and histamine release (13, 14). The present theory is that cytokines are responsible for many of the inflammatory changes induced by pathogenic organisms during OM (15).
Different cytokines are involved in the early and late stages of inflammation and it has been observed in a bilateral study of effusion that cytokine production and concentration can vary between the ears (16). IL-1 and TNF-α are early-response cytokines with IL-1 being a more potent activator than TNF-α (17). IL-1 may be more important than TNF-α in the elicitation stage of inflammation, while there is more of a need for TNF-α during recruitment and maintenance stages (18, 19). IL-1 and TNF-α from macrophages induce the expression of glycoprotein adhesion molecules on the surface of vascular endothelial cells which bind leukocytes so they can leave the circulation and enter the site of infection.
IL-1
IL-1 was originally identified as a lymphokine that was mitogenic for murine thymocytes (20). Now IL-1 is known to be produced by many different cells to regulate immune responses (21). IL-1 is one of the most active substances inducing bone resorption through osteoclast activation (22). This IL-1-mediated bone destruction is one of the clinical characteristics that signal the onset of chronic OM. Neutrophils are a major producer of IL-1β. IL-1 induces the production of IL-1 in neutrophils in a positive-feedback mechanism (23). IL-1 has been shown to stimulate the synthesis of TNF, IL-2, IL-6, and IL-8 (24-27). IL-1 is comprised of two principal 17kDa polypeptides, IL-1α and IL-1β. Genes found on chromosome 2 encode these two molecular species (28). They have the same biological activities and bind to the same receptor on cell surfaces (28, 29). Both IL-1α and IL-1β are derived by proteolytic cleavage of 33 kDa precursor molecules (28). IL-1α acts as a membrane-associated substance, whereas IL-1β is found free in the circulation (30).
IL-1 promotes the release of other cytokines and stimulates arachidonic acid metabolism in both cyclooxygenase and lipoxygenase pathways (21, 31). IL-1 is primarily synthesized by activated macrophages. In these, IL-1 production is stimulated by LPS and leukotrienes (28, 32, 33). TNF also stimulates synthesis of IL-1 (14). Platelet activating factor (PAF) can enhance IL-1 release by the production of leukotriene metabolites (34). IL-1β is mainly produced and released extracellularly by inflammatory cells such as macrophages/monocytes (35) and IL-1α is localized intracellularly or on the surface of such cells (36). IL-1 induces cell adhesion molecules (intercellular adhesion molecule-1 and vascular cell adhesion molecule-1) that in turn stimulate the migration of leukocytes (18, 37).
IL-lβ has been shown to play an important role in the pathogenesis of otitis media with effusion (OME). When introduced into the murine middle ear, endotoxin derived from Haemophilus influenzae produced middle ear effusion (MEE) with significantly higher levels of IL-1β than controls, and inoculation of IL-1β caused similar pathologic changes to that of endotoxin. It has been observed that IL-1β upregulates the activity of sodium-potassium-chloride cotransporters. Also, IL-1β suppresses epithelial Na+ channel-dependent fluid transport. This upregulation of cotransporters may be one of the important factors in the excess fluid accumulation that is observed in OM (38, 39).
Additionally, the administration of anti-IL-1 receptor antibodies with endotoxin reduced the incidence of MEE and the pathological changes of the middle ear mucosa (40). Viable pneumococci inoculated into the middle ear of chinchillas caused an increased concentration of IL-1β after 6 hr (41). In a guinea pig OM model induced with nonviable H. influenzae, Sato et al. (42) suggested that IL-1β and TNF-α were produced by middle ear mucosa in the early stage of OM, which caused subsequent inflammatory cell accumulation and further production of cytokines such as IL-8 and TNF-α in the late stage.
Barzilai et al. (43) showed that IL-1 levels were significantly higher in culture-positive versus culture-negative AOM. Levels of IL-1 significantly decreased on days 4-5 of antibiotic therapy whether the bacteria were eradicated or not. This is in contrast to measured levels of TNF-α, which are more closely correlated with the presence of bacteria (44). On the other hand, it has been shown that age is inversely related to the concentration or detection rate of IL-1β by enzyme-linked immunosorbent assay (ELISA) in the MEE of OME patients. Therefore, treatment that reduces IL-1β may be an effective adjuvant in the early stage of OM in children (45, 46).
IL-2
IL-2 is a 15.5 kDa glycoprotein synthesized principally by activated T cells, although activated B cells may also have the ability to produce small amounts of IL-2 (47). The gene of IL-2 is located on human chromosome 4q26. It induces the proliferation and differentiation of T-cells, B cells, NK cells, monocytes, and macrophages. Upon release, IL-2 is bound by T-cells that have been primed by exposure to their distinctive antigens. Helper T-cells stimulated by IL-2 secrete cytokines to stimulate other lymphocytes. Cytotoxic T-cells and natural killer cells stimulated by IL-2 also proliferate and secrete cytokines, IFN-γ, granulocyte-macrophage colony-stimulating factor (GM-CSF), and TNF-α which preferentially activate monocytes and macrophages. IL-2 reaches peak levels early in the inflammatory cascade, and levels quickly drop before there is any clinical evidence of inflammation. IL-2 is a key factor in determining the TH1 response of T-cells to promote a strong cell-mediated response. TH1 cells are responsible for orchestrating cellular immunity, while TH2 cells (stimulated by IL-4) provide signaling for humoral immunity. However, IL-2 was not detected in MEE of AOM patients by ELISA nor was it found in an experimental rat model of AOM induced by inoculation of Streptococcus pneumoniae type 3 and nontypable H. influenzae by reverse transcription-polymerase chain reaction (RT-PCR) (44, 48). On the contrary, IL-2 was detected in the MEE of the chronic OME patients by ELISA (45, 49). It is believed that the inflammatory up-regulation of IL-2 is balanced by the down-regulatory effects of IL-10. Smirnova et al. (50) reported that an imbalance in the production of these cytokines induces switching to the chronic stage. Excessive IL-2 production can cause humoral inflammatory processes and/or chronic cell-mediated processes whereas IL-2 deficiency promotes persistence of OME, which can result in chronic OME. From this, it may be deduced that the ongoing chronic inflammatory state associated with chronic OME may contribute different cell types, and thus different cytokines, than those with AOM.
Tumor necrosis factor (TNF)
TNF is a pair of cytokines originally called cachectin (TNF-α) and lymphotoxin (TNF-β). TNF-α is a polypeptide produced mainly by stimulated macrophages but made by wide variety of cells including fibroblasts, T cells and B cells (51). TNF-β is produced predominantly by lymphocytes and its active form is a 25 kDa glycosylated polypeptide (52). These two forms of TNF are 30% identical at the amino acid level (53).
Endothelial cells and epithelial cells all have the capacity to produce TNF. TNF induces the acute phase of the inflammatory response and release of other cytokines. TNF activates polymorphonuclear leukocytes, promotes fibroblast proliferation, inhibits vascular endothelial and B-lymphocyte proliferation, and stimulates cartilage and bone resorption (54). Thus, TNF has many of the same functions as IL-1. In fact, IL-1β and TNF-α induce each other's production and have a synergistic effect in vivo (55-57). TNF stimulates arachidonic acid metabolism, leading to the production of prostaglandins and leukotrienes that are important in the pathogenesis of OM (58, 59). TNF-α is a strong inducer of IL-1, IL-2, IL-6, IL-8, and endothelial adhesion molecules including intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) (60-63). TNF-α is chemotactic to neutrophils, monocytes, macrophages, and lymphocytes (17).
TNF-α was detected in human MEE and it is now apparent that TNF-α is one of the most significant inflammatory mediators in OM (64). Animal models of OM induced by S. pneumoniae and LPS have all demonstrated elevated levels of TNF-α in the early stage of OM. Ball et al. (65) indicated that TNF is a significant mediator of LPS-induced MEE in a rat model. Higher expression of TNF-α in human OM confers higher risks of subsequent episodes of OM, longer courses of OM, and chronicity of OM (66-68). The results of these studies suggest a potential therapeutic role with the clinical application of TNF inhibitors, such as TNF binding protein or inhibitor TNF soluble receptor, to control the symptoms of AOM or to prevent chronicity and recurrence of OM (65, 66).
IL-4, IL-5, IL-13, and GM-CSF
IL-4, a 20 kDa glycosylated polypeptide, is produced mainly by CD4+ T cells, but also by mast cells and basophils. It functions primarily as a growth factor for B cells and some types of T cells (14). IL-4 supports the proliferation of activated T cells and drives their differentiation to TH2 cells. These T cells support the production of antibodies in a humoral immune response. IL-4 stimulates B-cells to increase IgG and IgE isotypes (21). There are also anti-inflammatory effects that are stimulated by IL-4. It controls numerous molecular processes which cause deactivation of inflammatory macrophages, suppression of inflammation, down-regulation of the production and secretion of pro-inflammatory cytokines TNF-α, IL-1, and IL-8 (50). Although Melhus et al. (48) reported that they could not detect mRNA of IL-4 in the animal AOM model induced by S. pneumoniae and H. influenzae, Jang and Kim (49) could detect IL-4 in the MEE of chronic OME patients and suggested that elevation of TH2 cytokines, such as IL-4 and IL-6, might be a contributing factor in the persistence of OME with allergy. In addition, IgG production shows increases in relation to IL-4 prevalence in AOM in systemically immunized mice due to IL-4 having the effect of inducing B-cell differentiation into IgG-secreting plasma cells (69). It is thought that this is the pathway leading to inflammation in patients with allergic disorders causing OM. Smirnova et al. (50) reported that switching from acute to chronic inflammation can be induced by IL-4 due to its ability to up-regulate mannose receptor expression on activated macrophages. Activated macrophage fusion and giant multinucleate cell formation are promoted by the mannose receptor and create the cellular background for the onset of chronic inflammation.
IL-5 is produced by T lymphocytes as a glycoprotein with a Mr of 40-45 and is also involved in the TH2 pathway of inflammation (70). IL-5 functions to induce differentiation of eosinophils and augments proliferation of activated B-cells (14). IL-5 specifically acts on B-cells to stimulate and maintain IgA production (71). In chronic otitis media with effusion (COME) in systemically immunized mice, IL-5 positive cells were more numerous than IL-4 cells (69). This finding is consistent with the finding of increased local IgA production seen during chronic otitis media caused by allergy.
Recent findings by Iino et al. suggest that IL-5 may play a key role in the pathogenesis of eosinophilic otitis media (EOM), a form of infection often associated with bronchial asthma and/or allergic rhinitis. EOM is commonly seen in adults in contrast to AOM and OME which are more often diagnosed in children. The role of IL-5 in EOM can be linked to its chemotactic effect on eosinophils. EOM is characterized by having a markedly viscous or gelatinous effusion with a high eosinophil count and middle ear mucous membranes that are thickened with numerous eosinophils. The eosinophilic infiltration is thought to be due to locally produced IL-5. Iino et al. (72) showed that higher concentrations of IL-5 appeared in EOM middle ear effusions than in those of OME patients. This finding is further evidence of the role IL-5 has in EOM and OME patients.
IL-13 has also been implicated in the pathogenesis of OME. This inflammatory cytokine was found in 25.9% (7/27) of effusions in a study by Smirnova et al. (73) It was also found that both IL-4 and IL-13 had a significant positive correlation with the concentration of mucin in MEEs. It has been shown in an allergic airway inflammation model that suppress both IL-4 and IL-13 may be necessary to reduce mucus production and allergic inflammation (74). More research is needed to understand the functions of how IL-13 works in conjunction with IL-4 as a pro-inflammatory cytokine during OM.
GM-CSF has also been found to play a role in OM. GM-CSF promotes the activation and prolonged existence of eosinophils. Together with IL-5, it is possible that up-regulation of GM-CSF can cause eosinophil-mediated inflammation. In contrast, GM-CSF may also prove to be a down-regulator of allergic inflammation due to its ability to induce apoptosis in eosinophils (75). Further research is necessary to gain a better understanding of GM-CSF in the middle ear.
IL-6
IL-6 is a glycoprotein with a molecular weight of 25 kDa. IL-6 is produced by many cells, including helper T-cells, macrophages, mast cells, neutrophils, epithelial cells, and fibroblasts. It stimulates B cells to differentiate and is often upregulated following infection (76). Its main function is regulation of immune response, acute phase reaction, and hematopoiesis (77). IL-6 has been shown to play a key role in the acute phase reaction, including the induction of C-reactive protein (78). C-reactive protein is commonly used as a marker of bacterial infection. However, its use as a screening test for bacterial AOM is limited by its limited sensitivity and negative predictive value (79).
IL-6 seems to play an important role in the cytokine network of OM. The concentrations of IL-1, IL-6, and TNF-α in MEE from children were highly correlated with each other (80). Kerschner et al. (81) also discovered a significant correlation between MEE IL-6 concentrations and degree of hearing loss. In cultured middle ear epithelial cells, mucin secretion is upregulated by IL-6 in a dose and time-dependent manner. Interestingly, in a study of children with AOM, serum samples from the AOM patients infected with S. pneumoniae contained significantly higher levels of IL-6 than those infected with other bacteria or no bacteria (82). It appears that S. pneumoniae stimulates IL-6 production more than other types of bacteria. On the contrary, IL-6 was not detected in the guinea pig OM model induced by nonviable H. influenzae (41). Foglé-Ansson et al. (83) found that animals inoculated with gram-negative bacteria mediated a faster IL-6 response than animals inoculated with gram-positive bacteria. They also found that it was possible to determine whether an infection was grampositive or negative by analyzing the concentrations of IL-6. In a rat model, IL-6 mRNA increased in AOM induced by both S. pneumoniae and H. influenzae (48). Additionally, it was found that the shorter the course of OM, the higher the concentration of IL-6 in MEE (67).
In conclusion, expression of IL-6 is thought to be a potential candidate for the detection of the severity or chronicity of AOM. Further proof of this can be seen in the recent findings linking IL-6 to mucin upregulation. However, more reports demonstrating direct correlation between IL-6 and upregulation of mucin secretion are necessary to show transition to chronicity of OM.
IL-10
IL-10, known as cytokine synthesis inhibitory factor, is considered an immunosuppressive regulator of acute inflammation. It is produced by monocytes, CD4+ T cells, activated CD8+ T cells, and activated B cells following induction by bacterial products such as LPS. It is produced relatively late when compared to other cytokines, but there is evidence that IL-10 expression is upregulated shortly after or even simultaneously with pro-inflammatory cytokines. IL-10 is the principal TH2-type cytokine that upregulates humoral immune responses and attenuates cell-mediated immune reactions. If IL-10 is present for an extended period in the inflammation zone it can induce the amplification of chronic humoral inflammatory processes. Through this humoral inflammatory amplication IL-10 is thought to contribute to the switching of the infection into the chronic stage. A deficiency of IL-10 has also been seen to result in the chronic condition because the inflammatory actions of IL-1β, TNF-α, and IL-6 are not downregulated (50). IL-10, being the key anti-inflammatory cytokine, inhibits monocytes/macrophages and the production of TNF, IL-1, IL-6 and IL-8 (84). In addition, the production of oxygen radicals and proteases from activated neutrophils is directly suppressed. IL-10 was detected in the MEE of an experimental OM model by ELISA (85), and middle ear mucosal upregulation of its mRNA expression was also reported in the early and in the late stage of experimentally induced OM (48, 85). It is thought that IL-10 contributes to the eradication of middle ear inflammation and provides a negative feedback mechanism related to TNF-α in OM.
It is also believed that production of IL-10 must be balanced relative to production of IL-2. These cytokines need to be proportional in the middle ear in order to cancel the effects of the other. IL-10 is responsible for the down-regulation of acute inflammatory events while IL-2 leads to the up-regulation of cellular and molecular inflammation events (50).
Transforming growth factor (TGF)
TGF is divided into two classes of polypeptide growth factors. Transforming growth factor alpha (TGF-α) and beta (TGF-β) are not structurally or genetically related to one another and they act through different receptor mechanisms. TGF-β is known to exist in at least five subtypes: TGF-β1 through TGF-β5. TGF-β is part of a supergene family of low molecular weight cytokines that play a crucial role in the initiation and maturation of inflammatory processes including recruitment, activation, and proliferation of inflammatory cells (86). TGF-β has been found to stimulate extracellular matrix formation (fibronectin, collagen, and proteoglycans), protease inhibitor, and fibroblast chemotaxis. TGF-β is also believed to have an effect on fibroblast proliferation (87). The overproduction of TGF-β in the middle ear can elicit responses that may lead to chronic OME. Fibroblast accumulation and activation, IgA+ B cell secretion, and monocyte accumulation and activation are the result of TGF-β overproduction. In the presence of cytokines such as IL-10, IL-4, and GM-CSF, these effects can cause cell-mediated inflammation, humoral inflammation, and progressive fibrosis which leads to the chronic condition of OME (50). In a study by Cooter et al. (86), it was found that detectable amounts of TGF-β1 and TGF-β2 occurred in 100% and 98% of middle ear effusions, respectively. The major role of TGF-β occurs in the later stages of inflammation and its higher levels indicate that it participates in the repair of damaged tissue in the middle ear (50).
IL-12
IL-12, a heterodimeric pro-inflammatory cytokine, is produced by phagocytic cells such as macrophages, neutrophils, and Langerhans cells. When stimulated by bacteria or bacterial toxins such as LPS, these cells release large amounts of IL-12. This effect is further enhanced in the presence of interferon-γ (IFN-γ). IL-12 is a potent inducer of IFN-γ production by T and NK cells. IL-12-induced IFN-γ exerts positive feedback in inflammation by enhancing IL-12 production. Wang et al. (88) found that IL-2 upregulates the expression of IL-12 and increases the response of NK to IL-12. IL-12 is the major cytokine responsible for the differentiation of TH1 cells, which are producers of IFN-γ. The enhancing effect of IFN-γ on IL-12 production may represent a mechanism by which TH1 responses are maintained. IFN-γ activates macrophages and NK cells, and potentiates the proliferation of activated T cells. IL-12 is important in early immunity because it stimulates innate resistance and generates a TH1-type immune response. It is believed to form a link between innate and adaptive immunity. There have been no reports about the expression of IL-12 in OM until now. It is thought that the function of IL-12 in the middle ear is limited.
Chemokines
Chemokines are a family of structurally similar small proteins, whose major role is to stimulate leukocyte chemotaxis (89). In this way, they are thought to regulate the immune response. There are 2 families of chemokines, α-chemokine (CXC) and β-chemokine (CC) chemokines. CXC chemokines include IL-8 and are chemotactic to neutrophils. CC chemokines are chemotactic to monocytes and lymphocytes.
IL-8
IL-8 comes from monocytes, macrophages, fibroblasts, endothelial cells, and lymphocytes (90). It is produced on stimulation with IL-1β, TNF-α, IL-13, LPS, Concanavalin A, or viruses from the previously listed cells (5, 17, 91, 92). Since IL-8 expression can be controlled by the primary cytokines, IL-1β and TNF-α, it is considered to be a secondary cytokine in middle ear inflammation (73). IL-8 is a selective chemotaxic agent for neutrophils (93, 94). IL-8 induces neutrophil chemotaxis, activation and degranulation, surface expression of adhesion molecules, respiratory burst, and cytosolic calcium increase (93). IL-8 increases adhesion molecules for attachment and migration of neutrophils during acute inflammatory response (95). Since IL-8 is capable of inducing lysosomal enzyme release, IL-8 may play a key role in causing tissue damage, which leads to the prolongation of middle ear inflammation (96, 97). When directly injected into the middle ears of healthy mice, IL-8 causes more inflammatory changes than when S. pneumoniae is injected into mouse middle ears (98). IL-8 secretion is enhanced in both S. pneumoniae and H. influenzae provoked cases of OM (73).
Several authors detected IL-8 in human MEE. Transcripts of IL-8 were detected in 75% of both pediatric and adults MEEs (97). The mean level of IL-8 in MEE from children was higher than that of adults (99). The peak level of TNF-α and IL-1β in the MEE in a guinea pig OM model preceded the peak in IL-8 (41). This delay of the IL-8 peak level suggests that IL-8 is produced by inflammatory cells which accumulated via the influence of TNF-α and IL-1β.
Levels of IL-8 were closely correlated with the presence of bacteria and neutrophils in the MEE of OM. With initiation of antibiotic therapy, levels of IL-8 were reported to fall as bacteria were eradicated from the middle ear (100). IL-8 has also been shown to be predictive of the number of neutrophils in MEE (100-103).
Smirnova et al. (104) assessed the effects of IL-8 and other proinflammatory cyokines on goblet cells using human goblet cell line HT29-MTX which secretes two OME-related mucins, MUC5AC and MUC5B. They proposed that goblet cells are target cells for IL-8, TNF-α, and IL-1β and that IL-8 stimulates prolonged mucin secretion from goblet cells and may be involved in the maintenance of OM in the the chronic stage. Additionally, recurrence of AOM within 1 month was associated with high IL-8 levels in the initial middle ear fluid (101). The results of these studies suggest the possibility of using the IL-8 level as a prognostic factor or parameter of clinical outcomes.
Monocyte chemotactic protein-1 (MCP-1)
MCP-1 is expressed by endothelial cells, fibroblasts, and epithelial cells stimulated by either IL-1 or TNF (18). MCP-1 is chemotactic for both monocytes and lymphocytes in vitro (105, 106). Recently, MCP-1 has been shown to be chemotactic for monocytes and lymphocytes in vivo (107). MCP-1 expression alone does not activate these cells, but leads to enhanced inflammatory response upon treatment with other stimuli such as LPS (107). MCP-1 increases the adhesiveness of human monocytes toward endothelial cells by stimulating an increase in the surface expression and activation of adhesion molecules (108, 109). MCP-1 has a central position in monocyte trafficking and activation and has been implicated in diseases characterized by monocyte-rich infiltrates, including atherosclerosis, rheumatoid arthritis, and multiple sclerosis (110). MCP-1 is important in recruiting monocytes in chronic inflammation (17). MCP-1 has been detected in higher levels in MEEs with combined bacterial and viral infections (111). MCP-1 can be seen to upregulate the activation of inflammatory cells in the presence of LPS.
Macrophage inflammatory protein-1α (MIP-1α)
MIP-lα is released by monocytes, mast cells, endothelial cells, fibroblasts, epithelial cells, B cells, and T cells. It has potent monocyte and lymphocyte chemoattractant properties and induces release of histamine from basophils and mast cells (112, 113). MIP-1α was detected 9 of 11 cases of MEE (111). It is likely that MIP-1 plays an important role in the microenvironment of the middle ear, especially with respect to basophil function (111). This mediator has the potential to be important in acute and chronic OM as it is released by monocytes and induces histamine release and monocyte chemotaxis.
RANTES
Regulated upon Activation, Normal T-cell Expressed and Secreted (RANTES) is a CC chemokine that is chemotactic to monocytes and lymphocytes. RANTES was reported to be detectable in 94 (82%) of 114 effusions. The concentration of RANTES was positively correlated with the endotoxin content (114). In vitro, RANTES was expressed in middle ear epithelium in response to proinflammatory stimuli (TNF-α) in a dose dependent manner. Schousboe et al. (114) speculated that the expression of RANTES might explain the recruitment of monocytes in OME, possibly as a result of TNF-α-mediated endotoxin stimulation. On the other hand, Jang and Kim (115) demonstrated that RANTES and eosinophilic cationic protein (ECP) in MEE of OME with allergy were significantly correlated and significantly higher than controls. Their results suggest the allergic role of chemokines in the pathogenesis of OME.
Mast cells
Mast cells are the most numerous resident leukocytes in the middle ear. When activated, mast cells release preformed mediators, histamine and tryptase, and de novo synthesized mediators, leukotrienes and prostaglandins. Mast cells also release various cytokines including tumor necrosis factor and IL-1. During OM the mast cell population increases dramatically and mast cells will use Toll-like receptors and CD48 to directly interact with bacterial products. Ebmeyer et al. (116) showed that mast cells may be a major contributor to the early inflammatory response and may provide a link between infection and allergy in the generation of OM.
Histamine
Histamine causes vasodilation, increased vascular permeability, and edema of middle ear mucosa (117-119). Histamine is released by mast cells present in the middle ear upon activation by either complement or binding of antigen to IgE antibodies which stud the exterior surface of mast cells (120-122). Through its vasodilatory effects, histamine can cause mucociliary and eustachian tube dysfunction (123-126). Histamine levels and mast cell numbers were higher in adenoids of children with serous OM than in children with normal ears, suggesting a possible mechanism of eustachian tube obstruction secondary to histamine released from the adenoids (127, 128). In addition, middle ear mast cells were increased twofold in patients with chronic middle ear disease (129). Histamine levels were found to be higher in mucoid effusions than serous-type effusions (121, 129). Thus, histamine appears to be an important mediator influencing both acute and chronic OME.
Vascular endothelial growth factor (VEGF)
Another factor thought to be responsible for increased vascular permeability is VEGF. It is extremely potent, estimated to be 50,000 times as potent as histamine (130). Platelet-activating factor is 1,000-fold more potent than histamine, which may mean that the permeability effect of VEGF is approximately 50 times greater than PAF. Kim et al. (131) found that severe inflammatory cellular infiltration, subepithelial edema, and vascular dilatation occurred in Sprague Dawley rats that were exposed to 0.1 µg and 1.0 µg of recombinant vascular endothelial growth factor. It is thought that perhaps VEGF is primarily responsible for increased vascular permeability and that it is expressed in the presence of histamine (132).
Bradykinin/kallikrein-kinin system
The plasma kinin-forming system consists of three plasma proteins that interact in a complex manner when bound to negatively charged surfaces. These factors are factor XII (Hageman factor), prekallikrein and high molecular weight kininogen (HMWK). Factor XII, once converted to factor XIIa, converts plasma prekallikrein to kallikrein. Kallikrein enzymatically cleaves HMWK to liberate bradykinin. In serous OME, bradykinin enhances the vascular permeability in the middle ear mucosa, causing plasma to leak into the middle ear cleft (133). This plasma leakage characterizes serous effusions (134). Active plasma leakage into the middle ear occurs during the early phase of inflammation (133). The degree of plasma leakage is low in mucoid effusions and varied in serous effusions (133). Decreased plasma leakage into the middle ear in mucoid effusions may be explained by the consumption of the kallikrein system (134). Thus, bradykinin is an acute phase mediator, induced by the kallikrein-kinin system, which enhances vascular permeability in the middle ear leading to MEEs.
Platelet Activating Factor (PAF)
PAF is a membrane phospholipid metabolite released through the remodeling pathway upon stimulation (135). PAF, IL-1, and TNF can induce each other's release and also their own generation in self-generating positive feedback cycles (35). PAF is released from human neutrophils, platelets, eosinophils, macrophages, mast cells, and vascular endothelial cells. PAF synthesis is stimulated by PAF, LTB4, LTC4, and LTD4 (136, 137). Its activities include stimulation of arachidonic acid release and metabolism, increasing vascular permeability, and activation of neutrophils, monocytes, and macrophages (138, 139). PAF induces neutrophil adhesion to endothelial cells (137). In addition, PAF appears to play a major role in the production of MEE by increasing vascular permeability, stimulating epithelial secretory activity, and reducing the mucociliary activity of the middle ear causing eustachian tube dysfunction (140). By stimulating mucous glycoprotein release, PAF appears to be a mediator in the hypersecretion of mucus in the middle ear cavity and eustachian tube (141). PAF has been shown to impair mucociliary clearance function in a dose dependent manner (142). The disruption of the mucociliary clearance function may be partly responsible for chronic MEE. Levels of PAF in samples of human MEE have been reported to be in the range of 5.5 µg/mL in purulent otitis media, 7.6 µg/mL in mucoid otitis media, and 31.5 µg/mL in serous otitis media (143).
Arachidonic acid metabolites: prostaglandins and leukotrienes
Products derived from the metabolism of arachidonic acid (AA), also called eicosanoids, affect a variety of biologic processes, including inflammation and platelet function (Table 2). AA is derived from cell membrane phospholipids of certain cells in response to some stimulus which activates phospholipase A2 (PLA2). Possible stimuli are many and include antigen-antibody complexes, oxygen free radicals, bradykinin, and thrombin. Leukotrienes (LTs) and prostaglandins (PGs) are enzymatically formed from AA by lipoxygenase and cyclooxygenase respectively. Metabolites of AA have been identified in human MEE as well as in experimentally induced MEE in animals (58, 143, 144). Indomethacin or ibuprofen, an inhibitor of prostaglandin-forming cyclooxygenase, has been reported to be effective in reducing the accumulation of MEE or mucosal thickness in the animal OM model (145, 146). However, LTs seem to be more pro-inflammatory than PGs in otitis media, according to previous animal studies (58, 144).
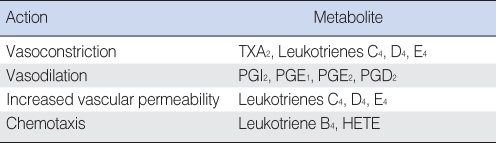
Inflammatory actions of eicosanoids
AA metabolites can affect the production of cytokines, and cytokines can regulate the production of AA metabolites. PLA2 activating protein stimulates PLA2 enzymes to break down arachidonic acid. The synthesis of PLA2 activating protein is induced in endothelial cells by IL-1, TNF, LTD4, and bradykinin (147, 148). Challenging the middle ear-eustachian tube (ME-ET) system with eicosanoids PGE1 and PGE2 resulted in increases in permeability and a vasodilation-edema. LTC4, LTD4, and PGE2 resulted in decreased ET patency and LTC4 slowed ME-ET clearance, suggesting that LT induces OME partly by impairing mucociliary transport of eustachian tube mucosa (117, 124, 149).
LTB4 is primarily synthesized by granulocytes and is chemotactic for neutrophils, monocytes, and lymphocytes (150). A study of experimental pneumococcal OM in chinchillas has suggested that arachidonic acid products, including LTs, in the middle ear are derived from inflammatory cells, since these products appeared after influx of inflammatory cells into the area (59). In animal models of OM, concentrations of LTB4, as well as other LTs, have been shown to correlate well with the degree of inflammation in the middle ear (146). LTB4 is higher in acute OM than chronic OM, suggesting it is related to the acute stage of middle ear disease (101). LTC4 was reported to be the most persistent eicosanoid in the infected chinchilla middle ear (151). Tada et al. (152) demonstrated that LTD4 can induce OME by a single inoculation and OME could last over 14 days, as LTD4 stimulated the release of proinflammatory cytokines. They also reported that oral administration of a specific LT antagonist, pranlukast, alleviated the experimental OME (152). Other LT-inhibitors, such as montelukast sodium and SCH/37224, have been shown to have some efficacy in prevention or treatment of OME experimentally or clinically (141, 153).
Free radical production in OM
Nitric oxide (NO) is a free radical generated by NO synthase in many cells and tissues including neurons, macrophages, neutrophils, endothelial cells, smooth muscle cells, lungs, and respiratory tract epithelial cells, including middle ear epithelial cells (154). In OM, it is largely responsible for vasodilation, increased vascular permeability, and production of mucoid effusions (155). NO synthesized by activated inflammatory cells regulates the functions of the other cells involved in the inflammatory process. Thus, NO may be considered a secondary mediator of inflammation produced by the middle ear epithelium in response to primary proinflammatory cytokines, such as IL-1β and TNF-α (156). Watanabe et al. (157) demonstrated that both in vivo and in vitro application of these cytokines resulted in increased iNOS in the middle ear epithelial cells of rats. Studies have demonstrated that inhibition of intracellular NO production blocks the hypersecretion of mucin in guinea pig tracheal epithelial cells stimulated by reactive oxygen species, TNF-α, PAF, and histamine (158-160). In rat middle ears injected with lipopolysaccharide, inhibition of intracellular NO release blocked mucin release (155). NO is also a likely mediator of osteoclastic resorption in COM (161). Recent work by Jeon et al. (162) suggests that NO may be a crucial signaling molecule that plays a role in nearly every step of OM pathophysiology. It is also suggested that the damaging effects of NO might be a result of its metabolite reactive nitrogen species which are products of the NO and reactive oxygen species reaction. In addition to NO, various other oxygen free radicals can directly damage middle ear epithelium. Both S. pneumoniae and neutrophils are known to produce highly reactive oxygen free radicals which have been shown to directly damage middle ear mucosa in OM (163).
Acute and chronic inflammatory mediators
IL-1 and TNF-α are early-response cytokines, although IL-1 appears to be a more potent activator than TNF (17). IL-1β concentrations were higher in purulent middle ear fluids than in serous or mucoid MEE (1, 164), and significantly higher concentrations were also found in patients with culture-positive than in those with culture-negative AOM (1, 43, 164). On the other hand, high concentrations of TNF-α in MEE were reported to be associated with a history of multiple placements of tympanostomy tubes (80). Cultured monocytes, which had differentiated to macrophages, had a markedly reduced LPS inducible IL-1, but not TNF-α secretion, suggesting that TNF-α may be involved in chronic otitis with differentiated macrophages and bacterial endotoxin retained in the middle ear cleft (165). MEE from ears with chronic OME have been assayed for endotoxin and TNF-α with 40% of ears having retained endotoxin. There was also a statistically significant correlation between both TNF-α and IL-1β with endotoxin levels (166). More recently, Hebda et al. (85) demonstrated that mucosal mRNAs for IL-6, IL-10, IFN-γ, TNF-α, MCP-1, and TGF-β were upregulated through the 16 weeks of follow-up in an experimentally induced OM. Taken together, endotoxin and upregulated TNF-α production are considered to be important factors for the persistence or chronicity of OME.
LTB4 and IL-8 are produced during acute infection of the middle ear and these PMN-related inflammatory substances may play an important role in delaying recovery or in recurrence of AOM (101). LTB4, LTC4, PGE2, and PAF appear to be important in the acute inflammatory process of OM (58, 144, 167). Further research is needed to gain a better understanding of the roles leukotrienes, prostaglandins, and platelet-activating factor play on each other and on the middle ear.
Inflammatory cells in acute and chronic OM
Neutrophils, monocytes, and macrophages are the major cell types observed in middle ear effusions (168, 169). Neutrophils are the predominant cells involved in early host response against OM during invasion of bacterial pathogens. Neutrophils have been shown to be associated with the pathogenesis of not only acute OM but also chronic OME in children (170). Lysozyme accumulation in MEE preceded significant inflammatory cell influx with lysozyme present at 6 hr and inflammatory cells present after 12 hr in MEE in H. influenzae-induced experimental OM (171). The predominant cell populations in the mucosa and MEEs in COME are mononuclear cells, macrophages, and lymphocytes, with plasma and mast cells also present (172-176). Lysozyme is a product of both PMNs and mononuclear inflammatory cells, with levels higher in purulent otitis media (POM) than in serous OM (177). Inflammatory cells recruited to the ME were able to set up a delayed inflammatory reaction in chronic OM (138).
In chronic otitis media with repeated episodes of acute inflammation, neutrophils and macrophages infiltrate with each episode (178). The number of macrophages continued to increase for up to four weeks after eustachian tube obstruction in chinchillas (177). As the inflammatory process enters the chronic phase of OM, there is a continuing shift in the population of infiltrating leukocytes toward increasing numbers of mononuclear cells.
Pediatric and adult otitis media
Most of the human studies discussed focus on children because adult OM occurs relatively infrequently. There are differences in the inflammatory mediators involved in child and adult OME. Higher levels of lysozyme, a nonspecific defense lysosomal enzyme against bacterial infection, were found in the effusions of children compared to adults (179). The early response cytokines IL-1β and TNF-α were present in higher percentages of effusions from children than adults (85% of children vs. 12% of adults for IL-1β and 85% of children vs. 8% of adults for TNF-α), suggesting that adult OM is associated with lower expression of the early response cytokines (46). These observed differences in OM between children and adults may be due to adults having more developed immunity to different pathogens.
Histopathology of acute and chronic OM
Acute inflammatory changes include vascular dilatation, submucosal edema, and infiltration of phagocytic cells into the middle ear. After the tissue damage has subsided, submucosal fibrosis, calcification, ossification, and hypertrophy of squamous epithelia of the tympanic membrane develops (178). Serous otitis media induced by eustachian tube obstruction in an animal model revealed histopathological findings characterized by hyperplasia of epithelium and edema of the subepithelial space resulting from increased vascular permeabiltiy. POM induced by inoculation of pneumococci into the middle ear showed histopathological changes characterized by metaplasia of the epithelial lining and goblet cells, widening of the subepithelial space with capillary dilatation and neutrophilic infiltration, and the appearance of lymphocytes and plasma cells (180).
Middle ear mucosal changes in patients with persistent OME show thickening of the pseudostratified epithelium with an increase in goblet cell density and pronounced basal cell hyperplasia (181). Goblet cell proliferation is commonly seen after toxic influence on respiratory epithelium (182). Under abnormal conditions, the flat nonciliated epithelium of the middle ear was transformed into pseudostratified ciliary epithelium with an increase in goblet cells (183). A study of 123 temporal bones with chronic OM showed fibrous thickening of the mucoperiosteum and granulation tissue principally, but subepithelial glandular formation was also a common finding (184). Influenza virus inoculated into the nose causes an increase in the frequency of goblet cells in the nasopharyngeal portion of the eustachian tube (185). Increased frequency of goblet cells was also induced by pneumococcal antigens in the bulla of guinea pigs (186). It has also been observed that the ratio of albumin to immunoglobulin can shed light on the stage of infection and further deduce the pathomechanics of infection (187). Many of these changes are caused, in part, by inflammatory mediators in the middle ear.
Bacterial endotoxin and LPS
A percentage of chronic OME may be related to retained bacterial antigens which produce chronic local stimulation of the immune system contributing to persistence of ME inflammation and effusion (188). Endotoxin is an integral component of the outer membrane of all gram-negative bacteria and consists of a complex of LPS and protein. The LPS is composed of a lipid portion (lipid A) linked by 3-deoxy-D-manno-octulosonic acid (KDO) to a heterogenous sugar polymer referred to as O-antigens (189). The gramnegative OM pathogens, nontypable H. influenzae (NTHi) and Moraxella catarrhalis, however, do not have O-specific polysaccharide antigens. Endotoxin from these bacteria is therefore referred to as being composed of lipooligosaccharide (LOS) and protein (190).
Middle ear effusions containing 100 ng/mL to 1 µg/mL of LPS and 25 to 100 ng/mL of LPS stimulated TNF-α production by cultured human monocytes (191). Therefore, monocytes and macrophages might be expected to be activated to produce inflammatory cytokines such as TNF-α, resulting in a chronic inflammatory response (192). LPS has been shown to induce production of IL-lβ, IL-6, and TNF-α and significant levels of IL-1β, IL-6, and TNF-α have been found in MEEs (45, 193, 194). LPS is a potent stimulus for the production of macrophage-derived TNF and IL-1 (195). Non-typeable H. influenzae LOS induced increases in cultured human middle ear epithelial (HMEE) cell expression of IL-1β, TNF-α, MIP-1β, IL-8, IL-6, and MCP-1 mRNA expression (196). Barrett et al. (197) demonstrated that LPS activated ICAM-1 receptors, IL-8, and NF-κB, a ubiquitous transcription factor complexed to its inhibitor within the cytoplasm in in vitro studies using rabbit middle ear epithelial cells. Endotoxin is reported to be present in a high percentage of middle ear effusions and even in nonpurulent MEEs (198-200). In an experiment examining the endotoxin levels in mucoid, serous, adult, and acute MEEs, endotoxin was found in 76.8% of the mucoid effusions, 55.6% of the serous effusions, 47.8% of effusions from adult patients, and 100% of acute effusions (201).
H. influenzae type b endotoxin and the outer cell wall of nontypable H. influenzae induced OM in guinea pigs (202, 203). Killed S. pneumoniae produced an inflammatory reaction consisting of epithelial metaplasia, increasing edema, and a PMN infiltrate in MEEs in a chinchilla model (204). H. influenzae and S. pneumoniae antigen was present in 26% and 21% of human effusion samples, respectively (205). Inoculating a higher concentration of LPS into the middle ear caused the development of a longer-term OME in the guinea pig (206). In a mouse model it was shown that the subepithelial space of middle ear mucosa was severely thickened with the infiltration of a large number of mononuclear cells induced by endotoxin (207). Bacterial endotoxin delayed mucociliary transport in the chinchilla ME, suggesting that bacterial components trapped in ME may sustain the inflammation and contribute to the chronicity of the MEE (208). MEE itself induces inflammatory reactions in the ME cavity in animals (120). Intratympanic inoculation of human MEEs induced OME in guinea pigs and reduced ciliary activity (209). Maeda et al. (207) found that endotoxin-induced OM resulted in mucoid MEEs and that eustachian tube dysfunction is essential to a serous MEE. Addition of endotoxin in vivo caused an increase in mucosal cell proliferation and the formation of epithelial effusion products (210, 211).
In view of the high incidence of endotoxin in MEE, a number of studies have focused on the cochlear effects of endotoxin to determine whether endotoxin can induce pathological changes, including hearing loss, after direct injection of the inner ear or subsequent to the injection of the ME with endotoxin from various sources (190, 212-214). When 1 mg/mL of endotoxin was applied onto the round window membrane in normal guinea pigs, elevation of auditory brain stem response (ABR) thresholds were reported (213). Demaria (215) showed that endotoxin shed by NTHi during experimental OM penetrates the inner ear and binds to both tissue components and inflammatory cells in both the middle and inner ear. Therefore, it is probable that endotoxin is a significant factor that can induce sensorineural hearing loss as a sequela of OM.
Transcription factor, toll-like receptor and cytokines
Among a variety of transcription regulators, NF-κB has been shown to play a critical role in regulating the expression of large numbers of genes including cytokines, chemokines, and other mediators involved in inflammatory responses (216). NF-κB is a dimeric transcription factor that is composed of p50 (NF-κB1) and p65 (RelA) subunits (216). NF-κB is rapidly activated by a large spectrum of chemically diverse agents and cellular stress conditions including bacterial LPS, microbial and viral pathogens, cytokines and growth factors (197, 217, 218). In resting cells, NF-κB is retained in the cytoplasm but enters the nucleus in response to various stimuli including bacterial infections. Activation of NF-κB is controlled by an inhibitory subunit, IκB, which retains NF-κB in the cytoplasm. The activation of NF-κB by a NF-κB translocation-dependent pathway requires sequential phosphorylation, ubiquitination, and degradation of IκB as well as consequent exposure of a nuclear localization signal on the NF-κB molecule. Multiple kinases have been shown to phosphorylate IκB at specific amino-terminal serine residues (219, 220). Phosphorylation of IκB by IκB kinase (IKK) pathways leads to the nuclear translocation of NF-κB which, in turn, activates expression of target genes in the nucleus. NTHi, a well-known pathogen of otitis media, strongly activates NF-κB in a cultured human middle ear epithlelial cell line (HMEEC-1) via NF-κB translocation-dependent and independent pathways. The NF-κB translocation-independent pathway involves activation of the MKK3/6-p38 mitogen-activated protein kinase (MAPK) pathway (221). It has also been shown that NF-κB is synergistically induced by the combination of TNF-α and NTHi (222).
In the host innate immune system, the surface epithelial cells recognize the invading bacteria by directly interacting with pathogen-associated molecular patterns on a variety of bacteria via Toll-like receptors (TLRs) expressed on the host (223). Of these, TLR2 has been well studied by researchers. TLR2 can respond to a variety of Gram-positive products, including peptidoglycan, lipoprotein, lipoteichoic acid, and lipoarabinomannan (224). TLR2 also plays an important role in the NTHi-induced NF-κB activation (221). Collectively, when NTHi binds TLR2, several key inflammatory mediators including IL-1β, IL-8, and TNF-α are upregulated by the activation of NF-κB (221).
On the other hand, the promoter regions of many of the TNF-α-regulated genes contain DNA binding sites for NF-κB. TNF-α also contributes to the activation of NF-κB. The interaction of TNF-α with its receptor has been shown to activate several signaling pathways, including a MAPK/extracellular signal-regulated kinase kinase 1 (MEKK-1)-dependent MAPK signaling pathway and a NF-κB-inducing kinase (NIK)-IKK-IκBα pathway (225). Watanabe et al. (222) showed that NTHi and TNF-α, when present together, synergistically induce NF-κB activation via two distinct pathways: a NF-κB translocation-dependent and an independent pathway in epithelial cell lines including HMEEC-1.
Mucous secretion and mucoid otitis media (MOM)
MOM is characterized by viscous fluid, high in mucin concentration, which accumulates in the ME cavity. Hypersecretion of mucous glycoprotein can be harmful to the mucociliary transport system (227). Mucocilary dysfunction in the tubotympanum plays an important role in the pathophysiology of OME and the rheological properties of MEE also play an important role in the process of recovery from OME (228, 229). Mucin, or mucus glycoprotein, is the major viscous component of MEE (230). There was a positive correlation between the viscoelasticity of MEE and goblet cell densities of middle ear mucosa (231). Mucociliary clearance is dependent on the rheological properties or viscoelasticity of MEE (232, 233). Mucociliary transport rates reduce when the viscosity of effusion is excessively high or low (233). Mucociliary dysfunction was suggested in the middle ears of children with OME caused by a decrease in the number of ciliated cells, hyperplasia of goblet cells, and an abnormal interaction of cilia and mucus that would fail to transport mucus, possibly causing mucus accumulation in the ME cavity (234).
Proinflammatory cytokines, such as TNF-α and IL-8, may play an important role in the pathogenesis of MOM. Mucoid effusions from human MEE samples contained higher levels of IL-8 than serous effusions (235, 236). TNF-α enhances PLA2 expression (237). PLA2, exogenously administrated, increased mucous glycoprotein secretion in cultured chinchilla middle ear epithelial cells (238). This suggests that PLA2 increases the release of arachidonic acid, since arachidonic acid metabolites are known to be involved in mucous glycoprotein production and secretion (159, 239, 240). TNF-α markedly increased the steady-state level of MUC2 mucin mRNA in ME epithelial cells of rats in a dose- and time-dependent manner (237). Inoculation of TNF-α into the ME cavity followed by ET obstruction stimulated mucous cell metaplasia hyperplasia in the ME cleft, accompanied by abundant mucin or mucin-like glycoprotein in the MEE (241). Concentrations of nitric oxide metabolites were higher in MOM than in serous OM (242). TNF-α was reported to stimulate secretion of mucin by guinea pig tracheal epithelial cells via mechanisms involving activation of NO synthase (NOS) and generation of NO (243). Therefore, it is conceivable that NO may also play an important role in the pathogenesis of MOM.
PAF also stimulates secretion of mucus glycoprotein mediated by arachidonic acid metabolites, especially metabolites of the lipoxygenase pathway via a PAF-receptor dependent mechanism (244), and inhibits mucociliary clearance of the eustachian tube (245). The expression of soluble adhesion molecules, such as intercellular adhesion molecule-1 (ICAM-1), is known to be upregulated at the cell surface in inflammatory disease. The levels of soluble ICAM-1 in mucoid effusions were reported to be significantly higher than those in serous effusions (246). More recently, Maeda et al. (207) demonstrated that additional administration of endotoxin was needed to produce mucoid MEE after the induction of serous MEE by ET blockage. Collectively, mucoid otitis media appears to be a more inflammatory and chronic form of OM than serous OM.
NTHi utilize the TGF-β-Smad signaling pathway together with the TLR2-MyD88-TAK1-NIK-IKKβ/γ-IκBα pathway to mediate NF-κB-dependent MUC2 mucin transcription in the epithelial cell lines including in the human middle ear epithelial cell line HMEEC-1 (247). Jono et al. (248) also showed that NTHi strongly induced up-regulation of MUC5AC mucin via activation of the Toll-like receptor 2-MyD88-dependent p38 pathway in the epithelial cell lines including HMEEC-1. Activation of TGF-β-Smad signaling, however, led to down-regulation of p38 by including MAPK phosphatase-1 (MKP-1), thereby acting as a negative regulator for MUC5AC induction (248).
The role of cytokines in cholesteatoma pathogenesis
Cholesteatomas are characterized by migration of keratinized squamous epithelium positioned in a fibrous stroma into the middle ear and mastoid cavity. This review has focused mainly on the associated inflammatory process seen in chronic otitis media and acquired cholesteatoma. Although the pathophysiology remains to be clearly elucidated, it is presumed to be multifactorial, as many theories have been proposed and investigated (249). Generally, it is thought that cholesteatomas form from a retraction pocket in the pars flaccida of the tympanic membrane (TM). Predisposing conditions causing TM invagination appear to be chronic eustachian tube dysfunction with negative middle ear pressure, repeated bacterial infections, or chronic otitis media. In many of these cases, this induction of cholesteatoma formation seems to be related to both internal molecular dysregulation and external stimuli in the form of pro-inflammatory cytokines, growth factors and/or bacterial toxins. Acquired cholesteatomas almost universally arise in the setting of inflammation and infection. Inflammatory granulation tissue always appears with invading epithelium in active human cholesteatomas (250) and experimental animal cholesteatomas (251). The inflammatory milieu that occurs in the setting of chronic otitis and cholesteatoma has been well delineated. As previously described, a large number of pro-inflammatory cytokines, prostaglandins, growth factors, and inflammatory cells have been identified in both chronic otitis media and cholesteatoma (249, 252-255).
The pathobiologic reason for the observed increase in proliferation in cholesteatoma tissue is not completely understood. Recent studies indicate a transcription factor, termed inhibitor of differentiation (Id1), is involved in the proliferation of cholesteatoma epithelium via an NF-κB/cyclin D1 dependent mechanism (6, 187). First, Id1 is upregulated in the rat middle ear mucosa following the infection with penumococcus (39). Second, upregulation of Id1 results in thickening of the middle ear mucosa with epithelial cell metaplasia. This is via the upregulation of cell cycle activity: the NF-κB/cyclin D1/cyclin-dependent kinases (Cdk2/4)/E2F/proliferating cell nuclear antigen (PCNA) signaling (Fig. 2). Phospholipase C-γ1 is the central molecule that activates the phospho-inositide second messenger signal transduction pathway. It is responsible for transducing the signals from the tyrosine kinase and epidermal growth factor receptors (EGFR). Increases in signal transduction from EGFRs result in a variety of cellular events including the induction of hyperplasia, growth, proliferative cytokeratin expression, and tumor invasiveness. Immunohistochemical and protein immunoblot studies showed that phospholipase C-γ1 is over-expressed across the matrix of cholesteatoma (256). In cholesteatoma, EGFR expression is not limited to the basal layer, as it is in normal squamous epithelia. Its expression is increased in the suprabasal strata (257, 258). 75% of cholesteatoma keratinocytes express EGFR as opposed to only 10% of normal and canal keratinocytes (259). Transforming growth factor-α, the ligand for EGFR is also expressed across all layers of the cholesteatoma matrix (258).
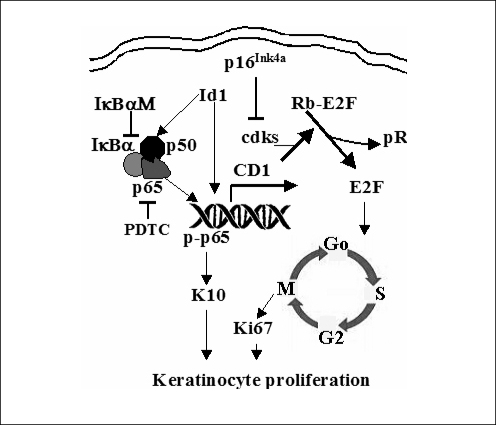
Schematic representation of the Id1-induced cellular hyperproliferation and abundant keratin 10 production pathways in keratinocytes. CD1: cyclin D1; Cdks: cyclin-dependent kinases (cdks 4/6); Rb: retinoblastoma; Rb-P: phosphorylated Rb; E2F: a transcription factor that drives the Go/G1-to-S phase transition of cells.
Although the molecular characteristics that contribute to cholesteatoma growth dysregulation have been qualitatively described, the exact pathobiologic mechanism leading to the observed hyper-proliferative state remains hypothetical. Evidence seems to point to the inability of cholesteatoma cells to abrogate proliferative and migratory signals from local inflammatory cells. Cytokines secreted by these local immune cells may create alterations in internal keratinocyte biochemical pathways that lead to the above-described hyper-proliferative phenotype. Moreover, noxious stimuli in the form of bacterial products and toxins could magnify this response. Ottaviani et al. (260) showed that the perimatrix of cholesteatoma showed a sharp reactivity to the ICAM-1 and to the endothelial leukocyte adhesion molecule (ELAM-1), both responsible for the homing of inflammatory cells at the epithelial-stromal junction of cholesteatoma. This over-expression of ICAM-1 and ELAM-1 seems to show that keratinocytes in cholesteatoma are in an activated state and that their hyperproliferation is mediated through inflammatory and/or autocrine cytokines. To this point, Huang et al. (261) has shown that GM-CSF, a protein that is highly expressed by monocytes and fibroblasts within the subepithelial stroma of cholesteatoma, is present at high levels throughout all layers of cholesteatoma. GM-CSF is known to induce in vitro keratinocyte proliferation and protein production.
It is theorized that intense infiltration of cholesteatoma by immune cells leads to chronic inflammation via continuous overproduction of certain cytokines. The reasons for failure to control this inflammatory response are not completely understood. It is possible that the microenvironment of the cholesteatoma state, namely granulation tissue, bacterial pathogens, and invasive hyper-proliferating keratinocytes, leads to a perpetual inflammatory milieu. Studies have shown the overall number of mast cells to be highly increased in acquired cholesteatoma (262). Mast cells are responsible for the secretion of a variety of potent cytokines many of which have been implicated in the pathogenesis of cholesteatoma. These include IL-1, IL-6, GM-CSF, interferongamma (IFN-γ) and TNF-α. Much like increased mast cell infiltration, acquired cholesteatomas contain increases in the number activated T-cells and macrophages (263).
Among the above-mentioned cytokines, TNF-α plays a major role in acquired cholesteatoma biology (264, 265). It appears to be preferentially localized to the connective tissue and epithelium of human cholesteatoma samples when compared to normal ear canal skin. In vitro, it has been shown to induce the proliferation, protein synthesis and terminal differentiation of basal keratinocytes. Although mainly secreted by activated macrophages, mast cells and cholesteatoma keratinocytes may also produce it. It induces osteoclast-mediated bone resorption (266), stimulates fibroblasts to secrete collagenase and prostaglandin E2 that in turn cause local soft tissue destruction (267), and may cause the resorption and inhibition of proteoglycans in cartilage (266-268). Furthermore, the levels of TNF-α in acquired cholesteatoma were correlated to the amount of inflammatory cell infiltration, bony destruction, and severity of infection (269). These facts all suggest that TNF-α plays a pivotal role in cholesteatoma pathogenesis.
Similarly, IL-1 was identified in the epidermis of cholesteatoma (270). IL-1 is produced both by cholesteatoma epithelial cells and by the inflammatory cells of the granulation tissue environment (252). Levels of both IL-1α and IL-1β appear to be much more elevated in cholesteatoma than in normal squamous epithelium (271). IL-1 has been implicated in the bony resorption process (272), and has been shown to stimulate the proliferation of keratinocytes (273).
Matrix metalloproteinases (MMPs) have been implicated as crucial agents in the development of bony erosion seen with cholesteatoma. MMPs are important in the physiologic turnover of the cholesteatoma epithelium extracellular matrix. Normally, their activity is tightly controlled, as an increase in their activation would cause denudement of the extracellular matrix and increased invasiveness by the epithelium. In cholesteatoma, studies have indicated a clear imbalance in the regulation of MMPs, with an overall upregulation of MMP expression and a decrease in MMP inhibitors resulting in degradation of the extracellular matrix. This causes the aggressiveness seen in cholesteatoma with regards to its invasiveness and bony destruction (274, 275). According to Wilmoth et al. (276), MMP2 levels were increased with exposure to LPS or TNF-α in cultured gerbil TMs. This supports a link between inflammatory mediators and the secretion of potentially destructive MMPs in the TM, which may contribute to the pathogenesis of cholesteatoma.
Interaction between middle and inner ear
Most of the middle ear inflammatory mediators and toxins from the middle ear enter into the inner ear through the round window membrane (277). It has been reported that the passage through the round window membrane (RWM) for small molecules is by passive diffusion and for large molecules by endocytosis (278). The blood-labyrinth barrier (BLB) also plays a primary role in the homeostasis of ion concentrations in the endolymph and periymph. Like other barrier systems, tight juctions within the capillaries of the spiral ligament form the morphological site of the BLB and prevent the passage of substances from blood into the fluid in the inner ear. Unlike other barrier systems, however, the BLB seems to be less permeable to several ions (sodium, chloride and calcium) and differentially modulates the passage of larger substances in proportion to molecular weight (Fig. 3) (279).

Pathological effects of inflammatory mediators in otitis media.
Ion channels, including the delayed rectifier K+ channel, Ca2+-dependent K+ channel, Ca2+ voltage-sensitive channel, and receptors such as N-metyl-D-aspartic acid (NMDA) and nicotinic acetylcholine receptors (nAchRs) have been localized to the stria vascularis, inner hair cells, and outer hair cells and are the primary factors responsible for maintaining the electrical potential of the hair cells and the process of auditory transduction (280). More than 75 different ion channels have been identified. Proper regulation of ion channel conductance is mediated by protein phosphorylation. Ion channel disorders such as tinnitus or metabolic disorders are characterized by a leakage in the apical or lateral potassium channel, affecting intracellular calcium levels.
CONCLUSIONS
Inflammatory mediators play a central role in the pathogenesis of OM and cholesteatoma by initiating and maintaining the inflammatory response to infection and injury. Inflammatory mediators may be one of the reasons that some patients progress from acute to COM and cholesteatoma. It is possible that inflammatory mediators such as histamine and PAF can lead to eustachian tube dysfunction resulting in decreased mucociliary clearance. This could lead to retained bacterial products such as lipopolysaccharide, cell walls, and bacterial antigens in the middle ear cleft causing a chronic inflammatory reaction with continual release of cytokines and arachidonic acid metabolites resulting in further inflammation and host cell damage. Another possible mechanism is through inflammatory mediators such as TNF-α, interleukin-l, and PAF induction of mucin secretion in the middle ear epithelium. This would cause increased viscosity of the middle ear effusion and may lead to decreased mucociliary clearance and retention of bacterial products causing chronic inflammatory reactions and mediator release.
Future studies are necessary to elucidate the roles of the newly discovered chemokines in the pathogenesis of OM and cholesteatoma. More studies are needed on the use of specific blocking agents to inflammatory mediators with the aim of discovering new treatment options for chronic otitis media and cholesteatoma. The future holds much promise in the field of inflammatory mediator research and through new discoveries the pain and suffering associated with otitis media can be reduced.
Notes
The authors declare that we have no conflicts in interests.