Growth Inhibitory Effect of Palatine Tonsil-derived Mesenchymal Stem Cells on Head and Neck Squamous Cell Carcinoma Cells
Article information
Abstract
Objectives
Mesenchymal stem cells (MSCs) play an important role in the development and growth of tumor cells. However, the effect of human MSCs on the growth of human tumors is not well understood. The purpose of this study is to confirm the growth effect of palatine tonsil-derived MSCs (TD-MSCs) on head and neck squamous cell carcinoma (HNSCC) cell lines and to elucidate the mechanism of their action.
Methods
TD-MSCs were isolated from patient with chronic tonsillitis and tonsillar hypertrophy. Two human HNSCC cell lines (PNUH-12 and SNU-899) were studied and cocultured with isolated palatine tonsil-derived MSC. The growth inhibitory effect of MSCs on HNSCC cell lines was tested through methylthiazolyldiphenyl-tetrazolium (MTT) assay. The apoptosis induction effect of MSCs on cell lines was assessed with flow cytometry and reverse transcriptase (RT)-PCR.
Results
Palatine tonsil-derived MSCs exhibited a growth inhibitory effect on both cell lines. Cell cycle analysis showed an accumulation of tumor cells predominantly in G0/G1 phase with an increase in concentration of TD-MSCs, which was confirmed by increased mRNA expression of cell cycle negative regulator p21. Apoptosis of tumor cells increased significantly as concentration of cocultured TD-MSCs increased. Additionally, mRNA expression of caspase 3 was upregulated with increased concentration of TD-MSCs.
Conclusion
TD-MSCs have a potential growth inhibitory effect on HNSCC cell lines in vitro by inducing apoptotic cell death and G1 phase arrest of cell lines.
INTRODUCTION
Mesenchymal stem cells (MSCs) are undifferentiated cells, derived from bone marrow, and widely distributed in various tissues of the body. MSCs can proliferate for many passages in culture and have the ability to give rise to several differentiated cell types under the influence of microenvironmental factors (1). These properties make them an attractive choice as a cell therapeutic agent (2-4). Growing evidences have shown that MSCs play an important role not only in regeneration and homeostasis, but also in the development and growth of human malignancies.
Sasser et al. (5) investigated the direct biological impact of primary human bone marrow stromal cells on the growth rates of a panel of metastatic breast cancer cell lines, and then found that human bone marrow stromal cells enhanced breast cancer cell growth rates in a cell line-dependent manner when evaluated in 3D tumor environments. In contrast to these tumor-promoting properties, Khakoo et al. (3) observed that intravenously injected-human MSCs homed to sites of tumorigenesis and potently inhibited tumor growth using an in vivo Kaposi's sarcoma animal model.
These findings means that the effect of MSCs on the tumor growth is not clear, and the inhibitory or promotive effect of MSCs on the tumor growth in vitro and in vivo is not well demonstrated (5-8). In addition, there are few reports for growth effect of MSCs on head and neck squamous cell carcinoma (HNSCC) cell lines.
Finding a suitable cell source of MSCs is a major challenge for cell therapy and tissue engineering. Although bone marrow (BM) has been the main source of MSCs (4, 9-15), the use of BM-derived cells is not always acceptable due to the high degree of viral exposure, the possibility of donor morbidity, and the significant decreases in cell number and proliferation/differentiation capacity with age (16). A highly invasive procedure is used to obtain BM, and, in this context, many efforts have been made to find an alternative MSC source in stem cell therapy. To date, MSCs have been isolated from a number of adult tissues, including trabecular bone (17), fat (18, 19), synovium (20, 21), skin (22), thymus (23), periodontal ligament (24) as well as prenatal and perinatal tissues such as umbilical cord blood (25), umbilical cord (26), and placenta (27).
Tonsils are easily accessible especially to otolaryngologist, particularly from young donors because of the prevalence of tonsillectomy procedure, and, if necessary, tonsillar tissue can be easily obtained by biopsy without major complications under local anesthesia. Therefore, we noticed that the tonsil can be another source of MSCs.
We performed this study to observe the influence of tonsil derived-mesenchymal stem cells (TD-MSCs) on growth of HNSCC and to elucidate the mechanism of the action further.
MATERIALS AND METHODS
Isolation and culture of tonsil stem cell
With institutional review board approval, tonsils were obtained after informed consent from patients undergoing tonsillectomy as a result of recurrent episodes of tonsillitis. To isolate tonsil stem cell, tonsil was washed extensively with equal volumes of phosphate-buffered saline (PBS), and tissue were digested at 37℃ for 30 minutes with 0.075% collagenase (type I; Sigma, St. Louis, MO, USA). Enzyme activity was neutralized with α-modified Eagle's medium (α-MEM), containing 10% fetal bovine serum (FBS) and centrifuged at 1,200 g for 10 minutes to obtain a pellet. The pellet was filtered through a 100-µm nylon mesh to remove cellular debris and incubated overnight at 37℃/5% CO2 in control medium (α-MEM, 10% FBS, 100 unit/mL of penicillin, 100 µg/mL of streptomycin). Following incubation, the plate was washed extensively with PBS to remove residual nonadherent cells. The resulting cell population was maintained at 37℃/5% CO2 in control media. One-week later, when the monolayer of adherent cells has reached confluence, cells were trypsinized (0.05% trysin-EDTA; Sigma) resuspended in α-MEM containing 10% FBS, and subcultured at a concentration of 2,000 cells/cm2.
Adipogenic, osteogenic, and chondrogenic differentiation of TD-MSCs
Adipogenic differentiation was induced by culturing tonsil stem cell for 2 weeks in adipogenic media (1 µM dexamethasone, 100 µg/mL 3-isobutyl-1 methylxanthine, 5 µg/mL insulin, and 60 µM indomethacine, 10% FBS in α-MEM) and assessed using an oil red O (Sigma) stain as indicator of intracellular lipid accumulation. Prior to staining, the cells were fixed 15 minutes at room temperature in 70% ethanol. The cells were incubated in 2% oil red O reagent for 1 hour at room temperature. Excess stain was removed by washing with 70% ethanol, followed by several changes of distilled water.
Osteogenic differentiation was induced by culturing ADSC for 2 weeks in osteogenic media (0.1 mM dexamethasone, 10 µM β-glycerophosphate, and 50 µg/mL ascorbic acid, 10% FBS in α-MEM) and examined for extracellular matrix calcification by alizarin red S (Sigma) staining. For alizarin red S staining, the cells fix with 70% ethanol and washed with distilled water. The cells were incubated in 2% arizarin red solution for 15 minutes at room temperature. The cells were washed several times with distilled water.
For chondrogenic differentiation, cells were stimulated in chondrogenesis induction media for 3 weeks. Cells were then rinsed twice with PBS and fixed in 10% formalin for 15 minutes at room temperature. Alcian Blue (Sigma) was used to stain chondrogenic pellets. Excess dye was removed, and cells were visualized by light microscopy.
Flow cytometry analysis
Flow cytometry analysis was used to characterize the phenotypes of the ADSC. At least 50,000 cells (in 100 µL PBS/0.5% BSA/2 mmol/L EDTA) were incubated with FITC-labeled monoclonal antibodies against human CD73, CD90, CD105, CD45, CD31, CD117, HLA-DR (BD Biosciences Clontech, Palo Alto, CA, USA) or with the respective isotype control. After washing the labeled cells were analyzed by flow cytometry using FACS-Calibur flow cytometer and the CellQuest Pro software (BD Biosciences).
Tumor cell lines
The laryngeal carcinoma cell line (SNU-899) was purchased from the Korean Cell Line Bank (28). PNUH-12 cells originating from hypopharyngeal squamous cell carcinoma were established at the Department of Otolaryngology, Pusan National University College of Medicine (29).
Growth inhibition of tonsil stem cells on tumor cells
Human tonsil stem cells were isolated and treated with mitomycin C (MMC) (50 µg/mL, Sigma) at 37℃ for one hour to block the proliferation. Then cells were washed twice with PBS and plated 0.1 mL onto 96-well plates at different number of cells. Tumor cells were suspended in RPMI-1640 medium and added into wells with or without stem cells. The ratios of stem cells to tumor cells were 1:20, 1:10, 1:5, 1:2, and cells were cocultured for 48 hours. Then the culture was analyzed by methylthiazolyl-diphenyl-tetrazolium (MTT) assay.
In MTT assay, MTT (0.5 mg/mL, pH 4.7; Sigma) was added 4 hours before the end of the culture time. The supernatant was removed, 150 µL DMSO was added and shaken for 5 minutes at room temperature. The absorbance at 570 nm was measured with Microplate Spectrophotometer (Molecular Devices, Sunnyvale, CA, USA). Inhibitory effect of stem cells on viability of tumor cells was expressed as follows: inhibitory rate (%)=(OD570 of tumor cells-OD570 of tumor with stem cells+OD570 of stem cells control)/OD570 of tumor cells×100%. The tumor cells originated from head and neck squamous cell carcinoma were cocultured with TD-MSCs at different cell number ratios for 48 hours. In the time-dependent experiment, the ratio of TD-MSCs to tumor cells was 1:20. TD-MSCs and tumor cells were cocultured for 24, 48, and 72 hours, respectively. The fibroblasts were purchased from the Korean Cell Line Bank (28) and treated by MMC at the same concentration. Then they were cocultured with tumor cells as the control group so that the influence of the portion of MMC treated TD-MSCs mixed with tumor cells should be evaluated.
Analysis of cell cycle and apoptosis
To determine the effect of tonsil stem cells on cell death and cell cycle distribution of tumor cells, 5×104 stem cells were seeded onto 6-well plate and incubated for 18 hours. Then culture medium was removed, the ratios of stem cells to tumor cells were 1:20, 1:10, 1:5, 1:2 and cocultured for 48 hours. Cells were harvested, washed twice with PBS and measured by flow cytometry analysis, reverse transcriptase (RT)-PCR, and real-time PCR assay. In flow cytometry analysis, 0.5 mL of PBS containing propidium iodide (5 mg/mL, BD Biosciences) was added to the culture before flow cytometry analysis (BD Biosciences). In RT-PCR analysis, total RNA was extracted from cucultured tumor cells using TRIzol regent. RNA (1 µg) was reverse transcribed into cDNA using a PCR cDNA synthesis kit (Clontech Laboratories Inc., Palo Alto, CA, USA). cDNA was used in the PCR normalized to β-actin gene. RNA (200 ng) was reverse transcribed and amplified using TaKaRa One-Step RT-PCR Kit (Takara Shuzo Co., Shiga, Japan). PCR was performed using the following primers: p21 sense, (5'-CCCGAGAACGGT GGAACT-3') and antisense (5'-AGAGGGCAGGCAGCGTAT-3'), product size: 493 bp; caspase 3 sense (5'-ATGTCATCTCGCTCTGGT-3') and antisense (5'-TCTGTTTCTTTGCGTGGA-3'), product size: 604 bp; and β-actin sense (5'-GCCATGTACGTAGCCATCCA-3') and antisense (5'-AACCGCTCATTGCCGATAGT-3'), product size: 373 bp. After 32 cycles of amplification, 10 µL of the reaction mix was removed, analyzed by 1.5% agarose gel electrophoresis and visualized by ethidium bromide staining.
Real time PCR amplification of the cDNAs was carried out with the reverse-transecribed RNA, primers p21, product size: 493 bp; caspase 3, product size: 604 bp; and β-actin, product size: 373 bp, using the Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA). Reactions were run on a LightCycler detection system (Roche Molecular Systems, Alameda, CA, USA).
Statistical analysis
All experimental data were expressed as mean±SD of at least three independent experiments performed in duplicate. The statistical analysis was performed with the Student's t-test or covariance analysis for statistical significance. Statistical significance was set at a P-value of less than 0.05. All analyses were carried out using SPSS ver. 15.0 (SPSS Inc., Chicago, IL, USA).
RESULTS
Confirmation of multilineage differentiation of TD-MSC
To characterize the TD-MSC population, the isolated cells were analyzed using flow cytometry analysis. Cells were analyzed for the expression of cell membrane protein markers: positive for CD73, CD90, and CD105, which are generally considered as markers of MSCs, but negative for the expression of hematopoietic markers such as CD45, CD31, and CD117 (Fig. 1A).

Characterization and differentiation of tonsil derived-mesenchymal stem cells (TD-MSCs) analyzed by flow cytometry and microscopy. (A) The mesenchymal stem cell (MSC)-specific markers CD73, CD90, and CD105 were expressed in TD-MSCs, whereas the hematopoietic stem-cell markers CD45, CD31, and CD117 were not. (B) TD-MSCs showed a fibroblast-like morphology and had the ability for adipogenic, osteogenic, and chondrogenic differentiation in differentiation induction medium. Differentiated cells were stained with oil red O, alizarin red S, and alcian blue.
TD-MSCs showed a fibroblast-like morphology and were plastic adherent under standard culture conditions (Fig. 1B). The differentiation potential for mesenchymal lineage was assessed by oil red O, alizarin red S, and alcian blue staining. TD-MSCs exhibited a characteristic of differentiation morphology in induction medium but not control medium (Fig. 1B). Two weeks after adipogenic induction, morphology of lipid droplets were observed and stained by oil red O. Under osteogenic culture conditions after 3 weeks, calcium deposits were detected by alizarin red S staining. Chondrogenic differentiation was revealed by alcian blue staining after 3 weeks of chondrocyte induction (Fig. 1B).
Growth inhibition of TD-MSCs on tumor cells
TD-MSCs exhibited a number-dependent growth inhibitory effect on different tumor cell lines (Fig. 2), but not the time-dependent effect (data not shown). However, fibroblast treated control showed neither a number-dependent nor the time-dependent growth inhibitory effect. In addition, TD-MSCs exhibited significant growth inhibitory effect on each tumor cell line more than fibroblasts at the cell ratio of 1:5 or 1:2 (Fig. 2).

Tonsil derived-mesenchymal stem cells (TD-MSCs) shows methylthiazolyldiphenyl-tetrazolium (MTT) metabolism inhibitory effect on head and neck squamous cell carcinoma (HNSCC) tumor cells by number-dependent manner. 5×104 cells of each head and neck cancer cell line (PNUH-12 and SNU 899) were incubated with mitomycin C (MMC)-treated TD-MSC or fibroblast at different cell ratio (1:20, 1:10, 1:5, 1:2), and cell viability was evaluated by the MTT assay (A) and (B). Results are mean inhibitory rate±SD of three independent experiments. *P<0.05 (compared with fibroblast treated control).
TD-MSCs induce cell cycle arrest of tumor cells in the G1 phase
We characterized the effect of TD-MSCs on the cell cycle of tumor cells to explain the mechanism of growth inhibition of MSCs. Cell cycle analysis showed an accumulation of tumor cells increased in G0/G1 phase with an increase in concentration of TD-MSCs, which was statistically significant (PNUH-12, 33.1 vs. 50.6; SNUH-899, 36.6% vs. 58.5%; P<0.05) on each tumor cell line (Fig. 3A, B). On the contrary, control showed the increased tumor cells in G0/G1 phase with an increase in concentration of TD-MSCs which was not statistically significant. Furthermore, the increase of G0/G1 phase was more in MSC treated SNU-899 cell line than in fibroblast treated control at the cell ratio of 1:5 and 1:2 (Fig. 3B). This phenomenon was confirmed by mRNA expression of cell cycle negative regulator p21. The results of RT-PCR demonstrated that TD-MSCs could upregulate mRNA expression of p21 in tumor cells significantly as concentration of TD-MSCs increased (Fig. 3C).
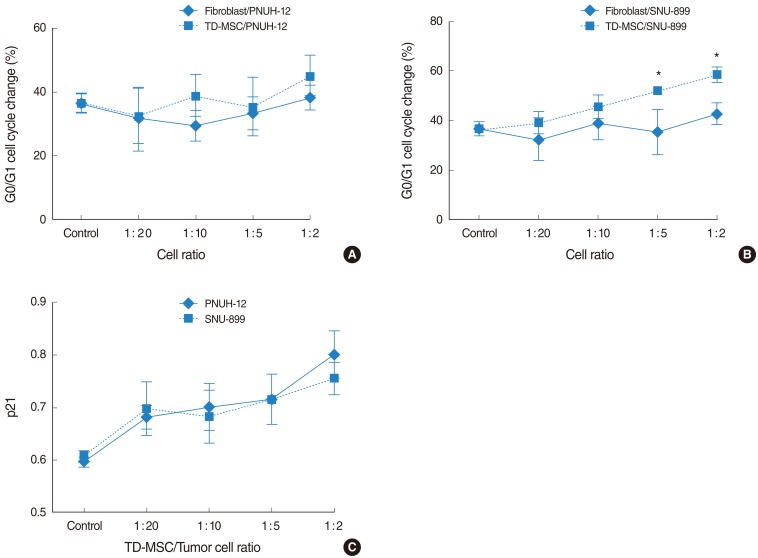
Tonsil derived-mesenchymal stem cells (TD-MSCs) induced cell cycle arrest of head and neck squamous cell carcinoma (HNSCC) tumor cells. 5×104 tumor cells were seeded onto 6-well plate and incubated for 18 hours. TD-MSCs and fibroblasts were cocultured for 48 hours at the cell ratio of 1:20, 1:10, 1:5, 1:2. Cells were harvested, washed twice with phosphate-buffered saline and measured by flow cytometry analysis. An accumulation of tumor cells increased in G0/G1 phase with an increase in concentration of TD-MSCs, which was statistically significant (PNUH-12, 33.1% vs. 50.6%; SNUH-899, 36.6% vs. 58.5%; P<0.05) on each tumor cell line (A) and (B). The increase of G0/G1 phase in TD-MSC treated SNU-899 cell line was more than in fibroblast treated control at the cell ratio of 1:5 and 1:2 (B). *P<0.05 (compared with fibroblast treated control).
TD-MSCs induce the apoptosis of tumor cells
To address this issue, HNSCC cells were cocultured with TD-MSCs, and tumor cells were harvested for apoptotic cell death and cell cycle distribution analysis. Apoptosis of tumor cells increased significantly as concentration of cocultured TD-MSCs increased on each tumor cell line (Fig. 4), and TD-MSCs exhibited significantly increased apoptosis of tumor cells more than fibroblasts at the cell ratio of 1:10, 1:5, and 1:2. Additionally, upregulated mRNA expression of caspase 3 was demonstrated with increased concentration of TD-MSCs (Fig. 5). TD-MSCs treated PNUH-12 cell line exhibited significantly increased mRNA expression of caspase 3 more than fibroblast treated control at the cell ratio of 1:2 (Fig. 5B).
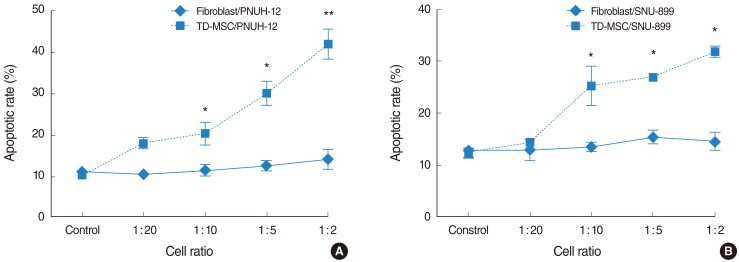
Cell cycle analysis shows apoptosis of tumor cells increased significantly as the cell ratio of cocultured tonsil derived-mesenchymal stem cells (TD-MSCs) increased on each tumor cell line (A) and (B). TD-MSCs exhibited significantly increased apoptosis of tumor cells more than fibroblasts at the cell ratio of 1:10, 1:5, and 1:2. *P<0.05, **P<0.001 (compared with fibroblast treated control).
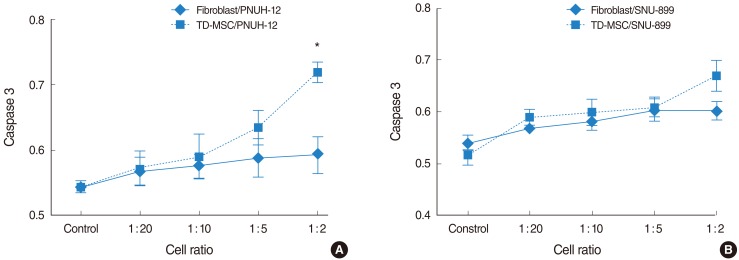
Upregulated mRNA expression of caspase 3 is demonstrated with increased concentration of tonsil derived-mesenchymal stem cells (TD-MSCs) at each tumor cell line (A) and (B), which could support the apoptosis of tumor cells. *P<0.05 (compared with fibroblast treated control).
DISCUSSION
MSCs were first characterized by Friedenstein et al. (11, 12), who identified an adherent, fibroblast-like population from adult BM. Over the ensuing decades, BM-MSCs have been characterized and have proven to have the capacity to differentiate into several mesenchymal lineages under appropriate conditions (4, 9, 11, 13). However, obtaining a BM sample requires some donor morbidities such as pain due to invasive procedure. These facts have led investigators to search for good substitutes for BM as an MSC source.
The practical application of MSCs is limited by ethical and logistic problems related to their isolation and their potential immunogenicity because of the requirement for allogeneic transplantation (2, 30-34). Ideal stem cells should be autologous cells that can be harvested without difficulty from the patient, manipulated efficiently in vitro, and implanted back into the same patient (32).
In this study, we isolated and cultured MSCs from palatine tonsil tissues, and showed that these MSCs were positive for cell surface markers of typical MSCs. Usually tonsil-derived MSC is a particularly interesting alternative source of MSCs because tonsil can be obtained with a less invasive procedure. Tonsillar epithelium is derived from the second pharyngeal pouch (of endodermal origin) and during fetal development is invaded by lymphoid tissue (of mesodermal origin). Therefore, embryologically, tonsils could be a source of MSCs. More importantly, the presence of MSCs in a secondary lymphoid organ underscores their potential contribution to a specialized microenvironment that supports the initiation and the maintenance of efficient immune responses.
Zhu et al. (8) reported that both fetal and adult BM could favor tumor growth in vitro (F6 or SW480) and in vivo. In melanoma murine tumor model, Djouad et al. (21) demonstrated the statistically higher incidence of tumor development in group injected with B16 tumor cells and MSCs than in the control group injected with the B16 tumor cells alone.
In contrast to these tumor-promoting properties, Lu et al. (35) investigated a number-dependent growth inhibitory effect of mouse BM-MSC on murine hepatoma and lymphoma cell lines and ascitogenous hepatoma cells in vivo without host immunosuppression. These finding could be explained by upregulated mRNA expression of cell cycle negative regulator p21 and apoptosis associated protease caspase 3 in tumor cells.
In addition, there are differences of the effect of MSCs on the tumor growth between in vitro and in vivo (3, 7). Ramasamy et al. (7) reported that MSCs exhibited a similar antiproliferative activity on tumor cells of hematopoietic and non hematopoietic origin in vitro, but when tumor cells were injected into nonobese diabetic-severe combined immunodeficient mice in conjunction with MSCs, their growth was much faster as compared to the group receiving only tumor cells. For the discrepancy between the in vitro and in vivo behavior, they suggested that MSC have the ability to form a cancer stem cell niche in which tumor cells can preserve the potential to proliferate and sustain the malignant process.
In this experimental study, MSCs were isolated from human palatine tonsil. Their phenotypes were positive for CD73, CD90 and negative for CD45. After cocultured with HNSCC cell lines, TD-MSCs exhibited growth inhibitory effect on tumor cell lines through decreased cell viability, which were explained by MTT assay. In addition, TD-MSCs exhibited a significant number-dependent growth inhibitory effect on tumor cells through increased cell cycle arrest and apoptosis. These phenomenon were supported by G0/G1 cell cycle changes and expression of p21 and caspase 3.
The current study demonstrate for the first time that TD-MSCs can induce potent growth inhibitory effect on HNSCC cell lines by increased cell cycle arrest and apoptosis. Their anticancer activities and mechanism of action, especially in vivo, deserve to be investigated further. The therapeutic application of MSCs still remain limited unless the favorable effect of MSCs for tumor growth in vivo and the long-term safety of the clinical applications of MSCs are better understood.
One of the limitations of this study is the possible influence of increasing portion of MMC treated TD-MSCs, which necessitate the application of MMC treated fibroblast for the control group. These factors originated from another limitation of the intermingled growth pattern between TD-MSCs and tumor cells. Notwithstanding these limitations, TD-MSCs showed the growth inhibitory effect on tumor cells that control group did not as the concentration of TD-MSCs and fibroblast increased. Moreover, TD-MSCs demonstrated the significantly increased growth inhibition and apoptosis of tumor cells more than control.
In conclusion, we isolated MSCs from human palatine tonsil successfully and demonstrated the growth inhibitory effect of TD-MSCs on HNSCC cell line by induction of cell cycle arrest and apoptosis.
ACKNOWLEDGMENTS
This study was supported by Medical Research Institute and Busan Cancer Center Grant (2010-27), Pusan National University Hospital.
Notes
No potential conflict of interest relevant to this article was reported.