INTRODUCTION
Approximately one in 1,000 children is affected by severe or profound hearing loss at birth or during early childhood, which is defined as pre-lingual deafness (1). About one half of childhood hearing loss is thought to have a genetic etiology, the majority of which is non-syndromic and not associated with abnormalities of other organ systems. Estimates from clinical and epidemiological studies suggest that 80-85% of hereditary, non-syndromic, pre-lingual deafness is autosomal recessive (1, 2).
Pendred syndrome phenotypes are associated with enlargement of the vestibular aqueduct (EVA) as detected by radiological imaging of the temporal bones (3).
We present a case of profound sensorineural hearing loss during early childhood with homozygous H723R mutation for SLC26A4 associated with bilateral EVAs.
CASE REPORT
The proband, a 3-yr-old boy, visited our clinic because of poor response to sound after head injury two months ago. After the accident, he became mute and timid, especially at his mother's absence. However, his mother reported he had been able to speak two-word combination and telegraphic sentences before head injury.
Otologic examination revealed normal-appearing and mobile tympanic membranes. No palpable goiter was present. And other findings of physical examination were unremarkable.
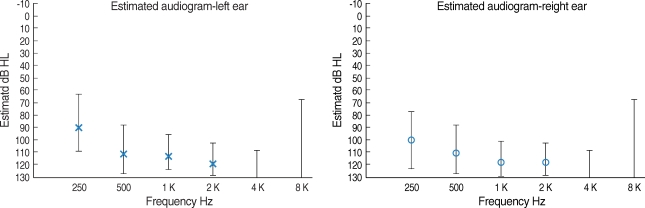
A sedated auditory brainstem response (ABR) revealed no identifiable wave V peak at 90 dB in both ears. And distortion-product otoacoustic emission (DPOAE) were absent in both ears. To obtain frequency-specific information and to check any residual hearing for fitting hearing aid, auditory steady state response (ASSR) was performed. ASSR showed significant response at high signal intensity (Fig. 1).
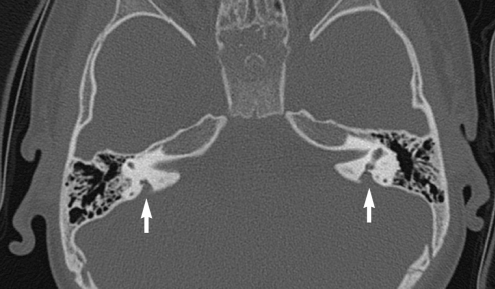
Temporal bone computed tomography showed bilateral EVA with no anomaly in the middle and inner ear (Fig. 2).
We recommended bilateral fitting of hearing aids and auditory rehabilitation prior to the decision to have cochlear implanation. Even though he was improved in the detection of sound and language with hearing aid, his performance was not satisfactory to follow the normal course of language development. After 6 months rehabilitation with hearing aids, cochlear implantation was done on his left side. Preoperatively, repeated ABR revealed no identifiable wave at 90 dB in the both ears. After 24 months of rehabilitation with bimodal binaural stimulation with CI and hearing aid, the hearing threshold in the free field was 27 dB A and the percentage of open-set monosyllabic word recognition was 92% in audio-visual (AV) condition and 64% in auditory only (AO) condition at 55 dB A.
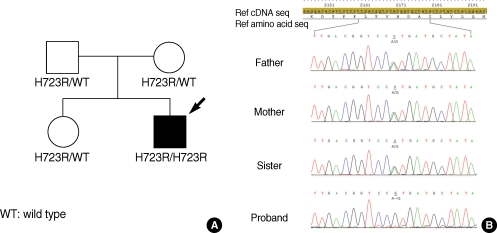
The genetic study showed H723R heterozygous mutation for all other family members, except the proband who showed H723R homozygous (Fig. 3). Thyroid function tests were performed to evaluate any possible thyroid dysfunction related with SLC26A4 mutation. The thyroid function tests showed thyroid-stimulating hormone (TSH) of 1.11 mU/L (normal range, 0.93 to 1.7), free triiodothyronine (FT3) of 4.0 pg/mL (normal range, 1.4 to 4.4), free thyroxine (FT4) of 1.84 ng/dL (normal range, 0.27 to 4.2), and thyroglobulin antigen of 33.6 ng/mL (normal range, 0 to 70).
Genetic analysis
Mutations were detected by a combination of intronic polymerase chain reaction (PCR) amplification, using primers flanking each exon, and sequencing of the amplification products. Exons 1-21 of the SLC26A4 gene were amplified from a genomic DNA samples by PCR. Amplification was carried out in a Perkin-Elmer thermal cycler using the following conditions: 5-min denaturation at 95℃, 37 three-step cycles (95℃ for 30 sec, 55℃ for 1 min, 72℃ for 1 or 3 min), 72℃ for 10 min, and ending with a holding period at 4℃. The PCR products were directly sequenced after removal of unincorporated dNTPs and primers by incubation of 50-100 ng PCR product at 37℃ for 30 min with 0.1 µL exonuclease I (Amersham Life Science, Cleveland, USA) and 1 µL shrimp alkaline phosphatase (American Life Science). The enzymes were heat-inactivated at 80℃ for 15 min. An aliquot of 6 pmol of either the forward or the reverse primer was used in standard cycle sequencing reactions with ABI Big Dye terminators, and run on an ABI 377 sequencer.
DISCUSSION
Overall, SLC26A4 mutations may account for between 5 and 10% of patients with prelingual hearing loss (4). The gene is located on chromosome 7q22-q31 and encodes a chloride-iodide transporter that is expressed in the thyroid, inner ear, and kidney. The gene has 21 exons that encode a 780 amino acid protein called pendrin, a 86 kDa glycoprotein with 11 or 12 transmembrane domains (5).
Each ethnic population has its own distinctive, diverse series of SLC26A4 mutant alleles which includes one or a few prevalent founder alleles (6). In Caucasoid populations, the mutations IVS8+1G>A, L236P, and T416P account for nearly half of all SLC26A4 mutation alleles (7). On the other hand, H723R, IVS7-2A>G, and IVS9+3A> Gare common alleles and account for the majority of SLC26A4 mutations in East Asian populations (6). Especially, the mutation H723R accounts for 40% of SLC26A4 mutation in Korean (8) and 53% in Japanese patients (9) and is the most frequently detected mutation in both Korean and Japanese populations, as a result of a common founder effect (6). The frequency of H723R heterozygote detection among normal Korean control is reported as 1.6% (6).
The Pendred syndrome is characterized by congenital sensorineural hearing loss and goiter (10). However, the infrequent detection of goiter may reflect reduced diagnostic sensitivity of manual palpation, or the young ages of the probands, considering the tendency for goiter in Pendred syndrome to begin around the time of puberty. Phenotypic variability has also been reported in two families carrying the same PDS missense mutation (11). Recent study reported that some missens pendrin product, hypofunctional SLC26A4 alleles, affected transport activity of pendrin (12). A modifier gene, hypo-functional SLC26A4 variants, or environmental factor (iodine uptake, nutrition, etc.) may contribute to such variability (9, 11, 12).
We successfully performed a cochlear implant on a patient with EVA. In the absence of syndromic features to guide genetic diagnosis, a detailed knowledge of the distribution of mutant alleles at specific loci for individual populations is required for more efficient diagnosis.