INTRODUCTION
Hereditary hemorrhagic telangiectasia (HHT) is an incurable progressive genetic disorder inherited in an autosomal dominant pattern that manifests as dysfunctions in vasculogenesis, angiogenesis and maintenance of the normal vasculature. Distinctions are made between small dermal and mucosal lesions called telangiectasias and visceral arteriovenous malformations, which are larger and mostly found in the liver, lungs and brain. In HHT, blood vessel malformations occur whereby dilation and deterioration of capillaries can result in a total loss of the capillary bed leading to a shunt between arteries and veins [1]. Since deformed vessels are less stable towards mechanical stress, epistaxis and visceral hemorrhage are cardinal symptoms of HHT together with visible telangiectasias on the skin and oral mucosa [2].
The worldwide prevalence of HHT is estimated at 1:5,000/ 8,000 and it is listed as an orphan disease by the U.S. Office of Rare Diseases (Genetic and Rare Diseases Information Center) and European Orphanet (classification number: ORPHA774). The incidence of HHT can be significantly higher in specific regions such as Cura├¦ao and Bonaire (1:1,331) or in the Haut-Jura region (1:2,351) of France [3]. In 2000, the Cura├¦ao criteria were introduced, which define the clinical diagnostic features of HHT as telangiectasia, recurrent epistaxis, visceral lesions, and a positive family history. If three criteria are met, diagnosis is regarded as confirmed [4,5]. Due to the progressive course of HHT, the application of these criteria is reduced in children. At present, there is no cure for HHT, and treatment is confined to iron supplementation and transfusions to treat anemia and blood loss and reducing the frequency of epistaxis by laser therapy or skin grafting [2,6,7].
Genetic testing of HHT patients permits a subclassification into HHT types 1ŌĆō5. The majority of HHT cases are HHT type 1 (HHT1; online mendelian inheritance in man [OMIM]: #187300) or type 2 (HHT2; OMIM: #600376), caused by mutations in the endoglin (ENG) or activin receptor-like kinase-1 (ACVRL1) genes, respectively [8]. Although the pathogenesis is similar, the rate of pulmonary and cerebral involvement may be higher in HHT1 [9]. Heterozygous mutations in ACVRL1 and ENG support a model of haploinsufficiency for HHT1 and HHT2 [6]. Mutations in ACVRL1 and ENG are globally relevant for the development of HHT in European, African and Asian populations [1,3,4].
Two loci on chromosomes 5 and 7 are associated with the development of HHT3 (OMIM: #601101) and HHT4 (OMIM: #610655), respectively [10,11], whereas HHT5 (OMIM: #615506) is caused by mutations in GDF2 on chromosome 10, coding for bone morphogenetic protein 9 (BMP9). Alterations in the SMAD family member 4 gene (SMAD4) can cause a combined syndrome of HHT and juvenile polyposis (OMIM: #175050) [12].
The ENG, ACVRL1 and SMAD4 proteins are all components of the transforming growth factor beta (TGF-╬▓) signaling pathway, which is pivotal for vascular development and endothelial cell maintenance [13]. The TGF-╬▓ superfamily is a group of regulatory proteins that includes TGF-╬▓ proteins and BMP proteins. In the canonical TGF-╬▓ pathway, signal transduction is performed by receptor complexes with serine/threonine kinase activity and SMAD transcription factors. Binding of a TGF-╬▓ ligand induces a type II TGF-╬▓ receptor (TGFBR2) dimer to recruit a type I TGF-╬▓ receptor (TGFBR1) dimer resulting in a heterotetrameric receptor complex, which leads to phosphorylation of a receptor-regulated-(R)SMAD protein, formation of an (R-)SMAD complex with a common mediator (Co-)SMAD and transport of the complex to the nucleus where transcription is modulated. In endothelial cells, ENG and ACVRL1 promote or inhibit angiogenesis depending on receptor complex composition. ACVRL1 serves as a TGFBR1, which in complex with a TGFBR2 mediates effects via R-SMAD1/5/8, resulting in the expression of genes associated with angiogenesis and cell proliferation.
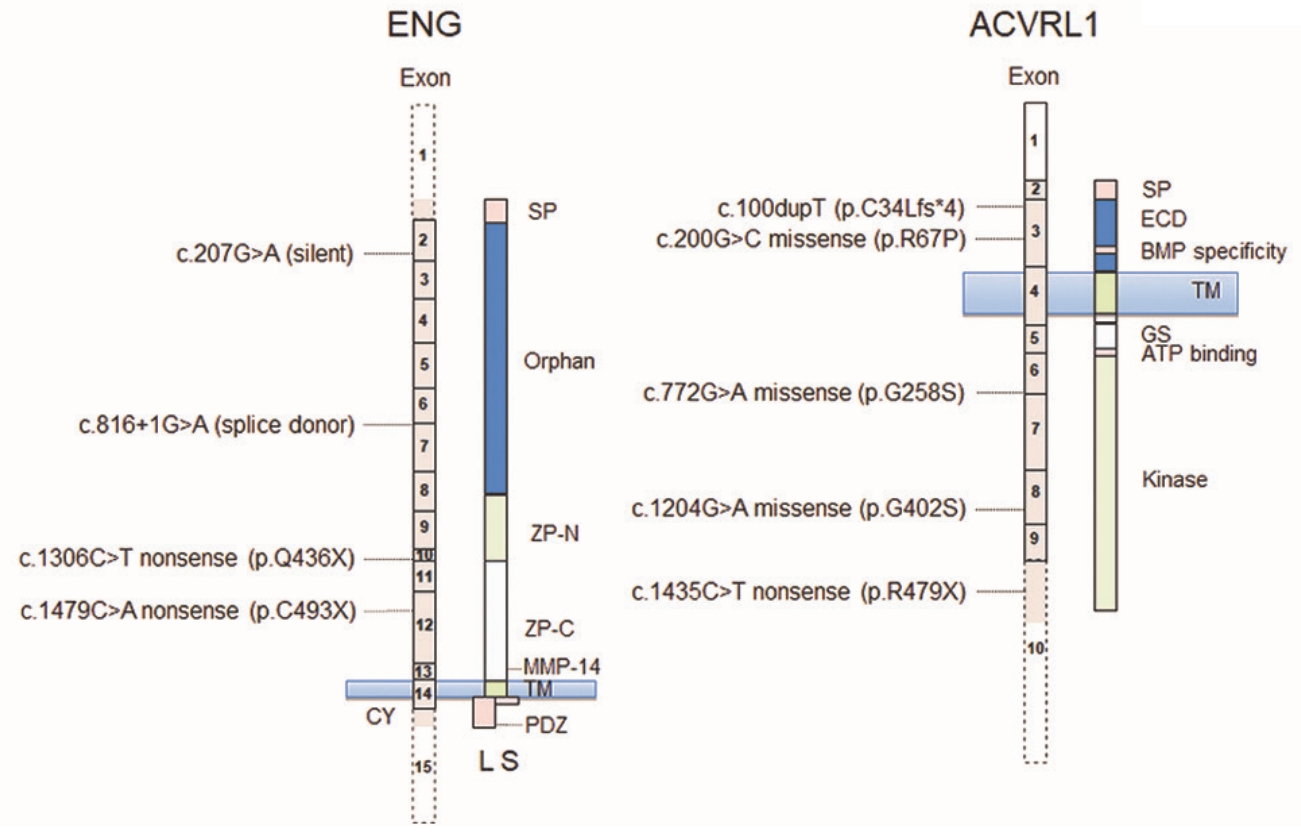
ACVRL1 is a 503-residue TGF-╬▓ type I serine-threonine kinase receptor with a single transmembrane domain expressed as a dimer on the surface of endothelial cells. Transcribed from 9 coding exons at 12q13.13, ACVRL1 has a 21-residue signal peptide and a short 96-residue cysteine-rich extracellular domain for interaction with TGF-╬▓ and BMP9 [14]. Residues within the extracellular domain govern specificity for BMP ligands [15]. ACVRL1 has an intracellular glycine- and serine-rich domain, phosphorylation of which by a type II receptor activates the C-terminal kinase domain to activate SMAD phosphorylation [13].
ENG is transcribed from 15 exons at 9q34.11 and encodes a 658-residue transmembrane glycoprotein expressed as a proangiogenic disulfide bridge-linked homodimer [16]. ENG has a large extracellular domain encompassing 586 amino acids that form two zona pellucida domains and an ŌĆ£orphanŌĆØ domain, which bears no homology to known proteins. The zona pellucida domain is thought to be important for dimerization, whereas the orphan domain seems to play a role in ligand recognition. The residues from position 1 to 26 serve as a signal peptide and are removed during processing. The ectodomain of ENG is highly glycosylated and is a dome-like structure of antiparallel arranged monomers shaped as a cavity [17]. The 47-residue serine-/threonine-rich intracellular region of L (large)-ENG contains a zona pellucida domain-binding motif that plays a role in modulating downstream signaling. Certain serine and threonine residues are constitutively phosphorylated. TGFBR1 and TGFBR2 interact with the cytoplasmic and zona pellucida-C domains of ENG [18]. A second isoform S-ENG (short) is generated by alternative splicing and has an intracellular region of only 14 residues [19]. Although ENG can bind some ligands such as BMP9 and BMP10 independently, it is primarily an auxiliary receptor that modulates signaling by affecting ligand binding in a complex with a TGFBR1 such as ACVRL1. A soluble form of ENG is generated by extracellular domain juxtamembrane metalloproteinase-14 cleavage. The soluble form of ENG can capture ligand [20] and is considered anti-angiogenic [13]. In this study, index patients from eight Austrian families diagnosed with HHT were screened for mutations in ENG and ACVRL1 to determine HHT1 and HHT2 status.
MATERIALS AND METHODS
Individuals suffering from HHT diagnosed according to Cura├¦ao criteria [5] were recruited from the Department of Otorhinolaryngology, University Hospital of Vienna. This study was approved by the Vienna Ethics Committee (No. 1328/2014) and compliant with the ethical standards due to the Declaration of Helsinki. Informed consent has been taken from any participant of this study prior to inclusion. Complete medical histories and otorhinolaryngological status of all individuals were examined. Clear autosomal dominant inheritance of HHT was observed in seven families.
DNA was extracted from peripheral blood with the Invisorb blood universal kit 1000 (STRATEC Molecular, Berlin, Germany). The full coding sequences of ENG (GenBank accession NM_001114753.2) and ACVRL1 (NM_000020.2) were amplified and sequenced as described previously with some modifications with primers [21] in a reaction mixture containing 200 ╬╝M dNTPs, 2 mM MgCl2, 20 mM Tris-HCl (pH 8), 50 mM KCl, 5% DMSO and 1U Platinum Taq polymerase (Invitrogen, Carlsbad, CA, USA) for 40 cycles of amplification with annealing at primer specific temperatures for 40 seconds, extension at 72┬░C for 60 seconds, denaturation at 94┬░C for 40 seconds and end extension at 72┬░C for 7 minutes in a GeneAmp PCR System 9700 thermal cycler (PE Applied Biosystems, Foster City, CA, USA). PCR products were separated on 1.5% agarose gels containing 0.5 ╬╝g/mL ethidium bromide at 1V/cm and sequenced on an ABI sequencer. Results were compared to the wild type locus sequence using the NCBI interface (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch&BLAST_SPEC=blast2seq&LINK_LOC=align2seq). Novel mutations were identified by literature searches and by reference to the ENG and ACVRL1 mutation databases at the University of Utah (http://arup.utah.edu/database/HHT). All identified variants were registered at ClinVar (SCV000346037-44). Sequences are ClustalW multiple sequence alignments (http://webcitation.org) with Homo sapiens (UniProt P37023), Pan troglodytes (H2Q5Y5), Canis lupus familiaris (E2R174), Mus musculus (Q91YV1), Rattus norvegicus (P80203), Gallus (XP_003643441.2), Xenopus tropicalis (F6XT30) and Danio rerio (Q498X0) sequences depicted with BoxShade (http://www.ch.embnet.org/software/BOX_form.html). The severity of missense mutations was predicted with Polyphen-2 (http://genetics.bwh.harvard.edu/pph2) and PROVEAN (http://provean.jcvi.org/index.php). Global allele frequencies were determined with the gnomAD browser (http://gnomad.broadinstitute.org) [22].
RESULTS
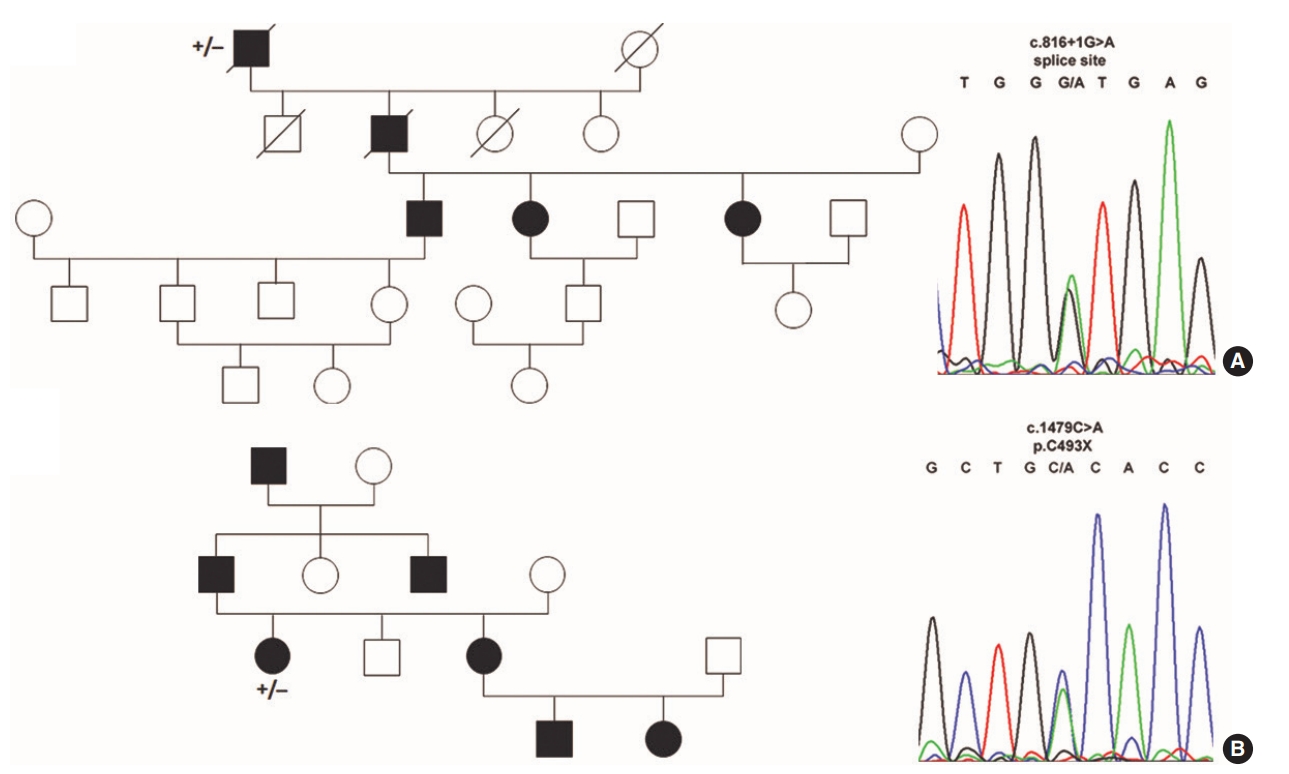
We identified three unpublished, five known and one silent variant in ENG and ACVRL1 (Fig. 1) in eight unrelated, nonconsanguineous families in Austria. In total, two novel variants and one known variant were identified in ENG. In one family (Fig. 2A) an unpublished intronic transition (rs111471193; ClinVar SCV000346037), listed in the ARUP database, was found (between exons 6 and 7; c.816+1G>A) that mutates the guanine residue in the highly conserved GU splice donor motif. A novel c.1479C>A nonsense p.Cys493X (SCV000346038) transversion in exon 12 was identified in a second family (Fig. 2B) causing the replacement of cysteine with an opal stop codon at p.493. The proband also suffered from a cerebral arteriovenous malformation, which was embolized with computed tomographic angiography. In the third family, a heterozygous c.1306C>T nonsense (p.Gln436X; SCV000346039) transition in exon 10 was found (Supplementary Fig. 1) that has been described previously [23].
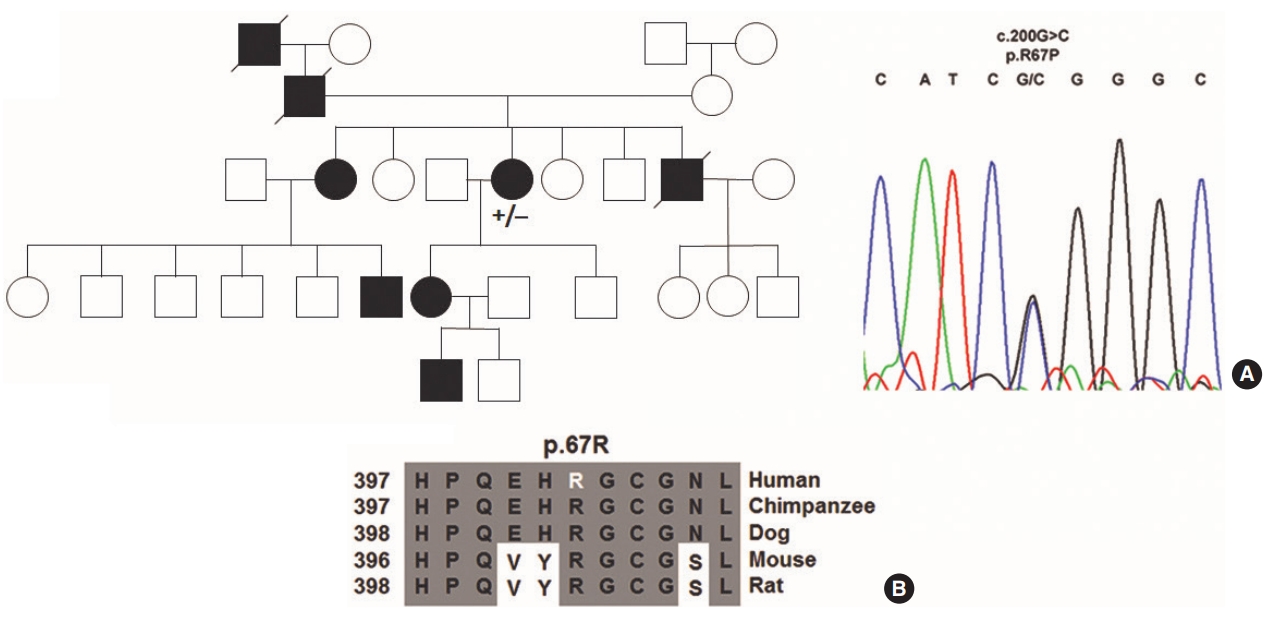
In total, one unpublished and three known variants were identified in ACVRL1. A heterozygous insertion of a thymidine after c.100dupT (Supplementary Fig. 2) in exon 3 (p.Cys34Leufs*4; HGMD CI111588; SCV000346042) was identified in one family that is listed in the ARUP database [24]. The index patient had pancreatic, hepatic and pulmonic arteriovenous malformations. A novel heterozygous c.200G>C missense transversion (p.Arg67Pro; SCV000346040) was found in the extracellular domain of ACVRL1 (Fig. 3) in another family. In the third family a c.772G>A (p.Gly258Ser; SCV000346041) missense transition in a highly conserved residue was found that is pending classification on the ARUP database. The mutation has been described previously [25] and is positioned at the distal end of exon 6 neighboring an atypical GC splice donor consensus (Supplementary Fig. 3). The index patient had pancreatic and hepatic arteriovenous malformations. Two other known variants found in two other families are the missense transition c.1204G>A (p.Gly402Ser; SCV000346043) [10,26] in exon 8 in a highly conserved residue (Supplementary Fig. 4) and the c.1435C>T (p.Arg479X; SCV000346044) nonsense transition in exon 10 (Supplementary Fig. 5) [27]. A single example of c.1435C>T is listed in the gnomAD database in a Latino subject (global 1/246,214; 4.1├Ś10ŌĆō6).
In addition to pathogenic variants, the c.207G>A transition in exon 2 of ENG was found in two families together with the known pathogenic ENG heterozygous nonsense transition c.1306C>T [23] (Supplementary Fig. 1) and the known ACVRL1 missense variant c.772G>A identified in this study (Supplementary Fig. 3).
DISCUSSION
In this study, nonsense, frameshift, splice donor and missense variants in conserved residues were identified. Late onset disease variants are commonly listed in allele frequency databases of healthy populations. The pathogenic variants identified are exceptionally rare and only a single listing of c.1435C>T worldwide is represented in the gnomAD allele frequency database [22]. Three of the mutations identified are unpublished in European and American populations to date and the pathogenic mechanisms leading to HHT1 and HHT2 can be predicted. Regardless of mutation, onset according to Cura├¦ao criteria was in early adolescence in all cases.
The ENG transition (c.816+1G>A) mutates the highly conserved GU intron 6 splice donor motif. If intron retention were to occur, translation from the mRNA would terminate at an opal stop codon at c.816+(88-90). The 29-residue peptide sequence ŌĆ£MSCAQLPGHKTQTPNLWIREVSWKGEPPSŌĆØ which has limited homology to a hypothetical putative enterobacterial transcription regulator DUF1323 (pfam07037) would be predicted to be translated distal to p.Trp272 within the orphan domain. A novel ENG variant identified is a nonsense c.1479C>A (p.Cys493X) variant predicted to cause synthesis of truncated ENG comprising only the N-terminal 437 residues [28]. It would therefore be expected that a translated peptide comprising the signal peptide, the orphan domain but only a fragment of the zona pellucida-C domain would be secreted, lacking the transmembrane and intracellular domains. Cysteine residues between p.330 and p.412 may cause dimerization between wildtype ENG and p.Cys493X ENG [28]. The truncated ENG would also not be subject to matrix MMP-14 cleavage in the juxtamembrane region of the zona pellucida-C domain at residues p.586-587 [19]. Amino acids within the zona pellucida-C domain between positions p.437-558 control interactions with TGFBR1 and TGFBR2, suggesting that p.Cys493X would disrupt TGF-╬▓ signaling modulation. This gene product, however, contains almost all domains present in a soluble form of ENG. Since the orphan domain is sufficient for binding BMP9/10, these ligands could be sequestered from their receptors by the mutant peptide to affect downstream pathways. Therefore, in addition to haploinsufficient effects caused by the absence of membrane bound ENG, p.Cys493X ENG could also lead to sequestration of circulating BMP.
The c.100dupT insertion in ACVRL1 (http://arup.utah.edu/database/HHT) is predicted to create an opal stop codon 11 residues after p.Thr33 causing translation to be terminated after 3 (ŌĆ£LHVŌĆØ) residues (p.Cys34Leufs*4). The novel ACVRL1 missense variant c.200G>C (p.Arg67Pro) is predicted by bioinformatics to be either possibly damaging (Polyphen-2-score, 0.95; sensitivity, 0.79; specificity, 0.95) or deleterious (PROVEAN: ŌĆō3.393). Variants in codon 67 have been linked previously to the development of HHT2. The c.200G>A (p.Arg67Gln) alteration [29] has been described previously [1] and c.199C>T (p.Arg67Trp) has been identified in an Italian family [30]. The extracellular domains of ACVRL1 and ACTRIIB have been crystallized in a complex with BMP9 (PDB;4FAO) [15]. The ACVRL1 extracellular domain (p.22-118) contains 5 ╬▓-pleated sheets (╬▓) numbered from the cleaved N-terminus (p.Arg67 is positioned in ╬▓4). Whereas the adjacent p.His66 residue forms a hydrophobic pocket at the BMP9 interface, the charged arginine residue at p.67 builds a salt bridge with p.Glu65 and charge-dependent interactions with the extracellular domain C-terminal residues p.Asn96, p.His97 and p.Val99. Replacement of cationic charged p.Arg67 with a neutral Pro residue could potentially interrupt these reactions similar to predictions for the uncharged glycine or tryptophan residue substitutions [15]. In addition, it has been reported that p.Arg67Gln is retained in the endoplasmatic reticulum [31].
Although silent, inheritance of the common c.207G>A (rs11545664; p.Leu69X) variant (gnomAD European non-Finnish allele frequency 0.104) in ENG may be weakly and nonsignificantly associated with sporadic brain arteriovenous malformations and reduces predicted binding scores for the splicing factor SRp40 [32]. In this study, the silent c.207G>A ENG variant was identified in patients (data not shown) also bearing the c.1306C>T ENG variant (no arteriovenous malformation development) and the c.772G>A ACVRL1 variant (pancreatic and hepatic arteriovenous malformation development).
In this Austrian study cohort, we determined the frequency of HHT1 and HHT2 as 37.5% and 62.5%, respectively. Expansion of the molecular diagnostic variant spectrum in HHT should accelerate the development of novel variant-specific therapies in the treatment of autosomal dominant HHT.