Incomplete partition (IP) type III is a rare genetic inner ear anomaly with distinct radiological features, including the absence of the modiolus and lamina cribrosa, and a corkscrew appearance due to a bulbous dilated internal auditory canal [1]. IP type III involves X-linked nonsyndromic hearing loss (DFNX), with mixed type and congenital fixation of the stapedial footplate, as well as the presence of a perilymphatic gusher due to the direct communication between the internal auditory canal and the cochlea [2].
DFNX is associated with six loci (DFNX1ŌĆō6), for which only five genes at five loci have been identified: PRPS1, POU3F4, SMPX, AIFM1, and COL4A6 for DFNX1, 2, 4, 5, and 6, respectively [3]. POU3F4 was the first gene identified in X-linked nonsyndromic hearing loss (NSHL) [4]. DFNX is characterized by profound mixed hearing loss, vestibular abnormalities, and congenital stapedial fixation with a perilymphatic gusher in males [2]. Computed tomography (CT) studies in patients with DFNX2 have shown abnormal dilatation of the internal acoustic canal and communication between the internal acoustic canal and the inner ear compartments. A subsequent molecular analysis revealed that the causative mutations were in POU3F4 (POU domain, class III transcription factor 4) [5].
The severity of the anomaly determines the outcomes of cochlear implantation (CI) in patients with cochlear deformities. For instance, the common cavity of the inner ear is a poor prognostic factor due to the difficulty in appropriately positioning the electrode inside the cochlea [6]. In IP type III, a cerebrospinal fluid (CSF) gusher displaces the electrode into the internal auditory canal, making CI challenging [7]. Moreover, the rarity of mutations in POU3F4 makes it difficult to predict the performance of CI [8-11]. Herein, we investigated the genetic predisposition of patients with IP type III and analyzed the performance of cochlear implants, with the goal of providing counseling guidelines for the treatment options, outcomes, and prognosis of DFNX2.
Eleven patients with IP type III based on radiological findings were enrolled between January 2005 and January 2022 at a single tertiary hospital. All the included patients had congenital NSHL without cytomegalovirus infection, middle ear infection, or ototoxic drug use. Auditory thresholds were evaluated using pure-tone audiograms or auditory brainstem responses. Inner ear anomalies were detected by a temporal bone CT. The categories of auditory performance (CAP) scores were used to evaluate the outcomes of CI in patients postoperatively at 3 months and 1 year.
For genetic testing, whole-exome sequencing or targeted sequencing was performed as described previously using the Agilent SureSelect V5 enrichment capture kit (Agilent Technologies) with sequencing on an Illumina HiSeq 2500 (101 bases paired-end) [12]. Copy number variations were analyzed as previously reported [13]. Subsequently, a multiplex ligation-dependent probe amplification assay identified large deletions and duplications.
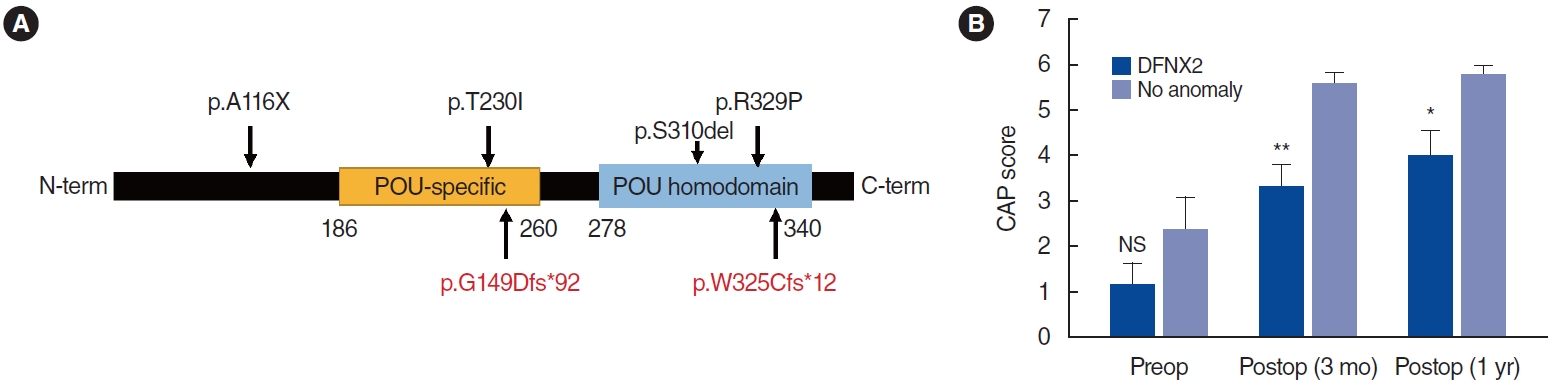
In nine out of 11 patients, pathogenic variants in POU3F4 were identified (Table 1) and two patients refused to undergo genetic testing. The variants comprised nonsense, missense, indels, and structural variants. Additionally, two novel frameshift (p.G149Dfs*92 and p.W325Cfs*12) variants were identified. The minor allele frequency in gnomAD was absent. According to the American College of Medical Genetics and Genomics/Association for Molecular Pathology guidelines, these mutations were likely pathogenic based on the PVS1, PM2, and PP4 criteria. p.G149Dfs*92 and p.W325Cfs*12 were located in the POU-specific domain and POU domain, respectively (Fig. 1A). The preoperative auditory thresholds were variable, while missense mutations (patients 1 and 7) or in-frame indels (patient 11) did not seem to be associated with more residual hearing than frameshift mutations (patients 2 and 8) or structural variations (patient 9) (Table 1). This finding suggests a similar level of loss of function regardless of the types of variants, though this finding should be interpreted with caution due to the small sample size of individual groups and the wide age range.
When CI was performed, all patients presented with a CSF gusher during the procedure, and a straight electrode ranging 16ŌĆō28 mm was inserted through the round window. As for the cochlear outcomes, the postoperative CAP score significantly improved from 1.2┬▒1.3 to 3.4┬▒1.1 (n=5, P=0.04) and 3.8┬▒1.5 (n=5, P=0.01) at 3 months and 1-year post-surgery, respectively. A comparison between the preoperative and postoperative CAP scores of patients with IP type III and those without a cochlear anomaly (n=5, age-matched) showed a statistically significant difference (Fig. 1B). In multivariable linear regression analysis that included age, sex, preoperative auditory threshold, other medical diseases, and genetic diagnosis, we could not find any significant factors to predict the outcomes (data not shown), which may be attributable to a small sample size of the patients. Although several patients (patients 3ŌĆō6) underwent CI surgery at an older age and might have had confounding factors, such as a longer duration of hearing aid usage, these results were consistent with the previous literature, which contains long-term follow-up data revealing poorer performance of CI in patients with IP type III than in a control group with GJB2 mutations [18,19]. These data indicate that patients with IP type III may have a limited number of auditory neurons and a suboptimal structure for electrode insertion and stimulation, resulting in restricted long-term auditory performance.
In the literature, patients with truncation or deletion variants tend to have poorer speech performance compared to patients without them [19]. However, we could not observe this tendency, since the CAP score at 1-year post-surgery was 2 in patient 1 with a p.T230I missense variant, while it was 6 in patient 2 with a p.G149DfsTer92 truncating variant. It is speculated that various clinical factors influencing the outcomes of cochlear implant may override the effect of a genetic deficit of POU3F4, which may cause variation in the surgical outcomes. Further research with a larger sample size cohort will be required to reach a conclusion about the genotype-phenotype correlation in terms of the surgical outcome of CI.
In summary, we identified POU3F4 genetic variants in patients with IP type III. There was no genotype-phenotype correlation depending on the type of variant, suggesting loss of function. Although the short-term CI performance was comparable to that in patients without a cochlear anomaly, the final performance during long-term follow-up may be poorer. These results should be considered during counseling and aural rehabilitation after surgery.