INTRODUCTION
Up to now over 400 syndromic forms of hearing loss (HL) have been described [1]. Pendred syndrome (PS) (MIM#274600) is one of the most frequent types of syndromic HL, although it may remain underdiagnosed due to the late onset (usually after the second decade of life) and reduced penetrance of some of its common clinical features, especially the goiter. Over 90% of PS is caused by SLC26A4 mutations in different populations [2].
SLC26A4 has been known by Everett et al. [3] using positional cloning and mapped on 7q22-31.1. This gene produces a 5 Kb transcript and a 86 kDa protein with 780 amino acids, called pendrin, which is a member of solute carrier family 26A (SLC26A).
SLC26A4 mutations result in both PS and non syndromic HL (DFNB4; MIM#600791). These disorders have autosomal recessive inheritance [2,4]. Similar HL severity and inner ear malformations ranging from enlarged vestibular aqueduct (EVA), the most frequent radiological abnormality in sensorineural HL (SN-HL), to Mondini dysplasia (MD), a condition that normal cochlear spiral is 1.5 turns instead of 2.5 turns, are seen in both of them. EVA and MD are detectable with computed tomography (CT) [5]. The major difference between the two disorders is thyroid abnormality (goiter) which is observed to be segregating with PS [2]. To date, more than 200 mutations have been identified for SLC26A4 (http://www.healthcare.uiowa.edu/labs/pendredandbor/slcMutations.htm).
While GJB2 is responsible for up to half of autosomal recessive nonsyndromic HL (ARNSHL) in Mediterranean, most European and north American populations [6-8], this gene accounts for only 18.29% and/or 16.7% of Iranian families with HL [9, 10]. In contrast, SLC26A4 appears to have a more crucial role in the etiology of hearing impairment (HI) in Asian than non-Asian populations. About 7.2% of prelingual HL in Pakistan and 5% of this disease in east and south Asia happens due to SLC26A4 mutations [11,12]. Results of a former study suggest that SLC26A4 could be considered as the second cause of HI, after GJB2, in Iran [13].
In addition to frequency, the type of SLC26A4 mutations is also different among populations. While IVS7-2A>G and H723R are the most frequent SLC26A4 mutations in east Asia, R79X and R409H are more common in the Iranian HL population [13-15]. Epidemiological data regarding ethnic and population specificity of SLC26A4 mutations could be beneficial for diagnosis, clinical decision-making, family planning as well as designing cost-effective strategy for molecular testing.
In the present investigation, a large deaf Iranian kindred with 9 patients was subject to genetic linkage analysis and two novel mutations were identified.
MATERIALS AND METHODS
Subjects
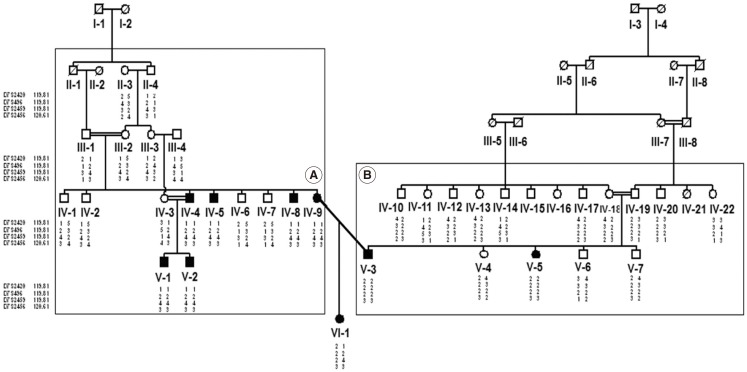
The research was approved by the Institutional Review Boards of Shahrekord University of Medical Sciences. We studied a large consanguineous family Iranian 3 (IR3), including 9 patients with hereditary HL from Fars province of Iran, with no GJB2 mutation in our previous research [9]. There were four consanguinity loops in the pedigree: subjects III-1 and III-2, IV-3 and IV-4, IV-18 and IV-19, and individuals III-7 and III-8 had first cousin marriages (Fig. 1). We also recruited 100 healthy matched controls from the same ethnic background. Written informed consent was obtained from all the subjects or their parents.
Phenotype evaluation
Audiological testing
For measurement of hearing, all patients underwent pure tone audiometry (PTA) test for air conduction at a range of frequencies from 250 to 8,000 Hz. Audiogram results of all patients were defined using a PTA average at three frequencies (500, 1,000, 2,000 Hz): mild for 21-40 dB, moderate for 41-70 dB, severe for 71-95 dB and profound for >95 dB.
Thyroid phenotype testing
The phenotype of thyroid was investigated with evaluation of its structure, size and function, based on sex and age of patients. Ultrasonography with Sonoline G50 ultrasound system (Siemens Medical Solutions, Erlangen, Germany) was applied to identify thyroid size and structure for all patients. For functional analysis of thyroid in all patients, thyroid stimulated hormone (TSH), thyroxin (T4), and triiodothyronine (T3) levels were measured by means of a chemiluminescent immunoassay (Berthold Technology-CSA, Bad Wildbad, Germany).
CT scan of temporal bone
All patients except V-5 and V-2 underwent high resolution CT scan by Somatom Sensation Emotion 16-Slice Configuration (Siemens Medical Solutions) for examination of vestibular aqueduct. EVA was defined as the diameter at the midway between the common crus and the external aperture being equal or more than 1.5 mm [16].
Molecular studies
DNA extraction
Genomic DNA for all available members of the pedigree as well as 100 ethnically matched normal control persons was prepared from 500 µL of peripheral blood by a standard phenol chloroform protocol [17]. For evaluation of quantity and quality of extracted DNA, spectrophotometry (UNICO 2100, West Springfield, MA, USA) and agarose gel electrophoresis were carried out according to routine methods.
Slink calculation, DFNB4 STR marker genotyping and linkage analysis
Selection of four short tandem repeat (STR) markers (D7S2456, D7S2459, D7S496, and D7S2420) and their primers were based on their physical distance, available at NCBI UniSTS and NCBI map viewer (http://www.ncbi.nlm.nih.gov/mapview).
Slink value was obtained by FastSlink ver. 2.51 option of Easy-Linkage ver. 5.05 [18]. Polymerase chain reaction (PCR) amplification of STR markers was performed as follow: 1 µL of MgCl2 (50 mM), 2.5 µL of Taq PCR buffer (10X), 0.4 µL of each of the primers (10 pM), 0.5 µL of dNTP mix (10 mM), 0.1 µL Taq DNA polymerase (5 U/µL), 2 µL DNA (50 ng), adjusted to 25 µL using ddH2O. For various primers some modifications were applied.
For amplification, the following touch-down PCR program was used: an initial denaturation at 95℃ for 5 minutes, followed by six cycles of 95℃ for 50 seconds (denaturation), 58℃ for 50 seconds in the first cycle with 1℃ reduction per cycle (annealing), and 72℃ for 50 seconds (extension) and 32 cycles of 95℃ for 50 seconds (denaturation), 53℃ for 50 seconds (annealing), 72℃ for 50 seconds (extension) and a final extension at 72℃ for 8 minutes. PCR products were loaded on a 14% polyacrylamide gel electrophoresis (PAGE) and run at 28 mA for 2-8 hours. Silver staining was performed to visualize the bands on the gel following standard protocols.
Two-point and multi-point logarithm of odds (LOD) scores were calculated by SuperLink ver. 1.6), and SimWalk ver. 2.91 options of EasyLinkage ver. 5.05, respectively [18,19]. STR markers haplotypes were reconstructed by SimWalk and visualized using Haplopainter ver. 029.5 software after linkage analysis [20]. Autosomal recessive (AR) mode of inheritance, complete penetrance, disease-allele frequency of 0.001, existence of no phenocopy, equal allele frequencies for markers and identical meiotic recombination frequencies in both sexes were assumed for LOD score calculations.
Mutation screening of SLC26A4
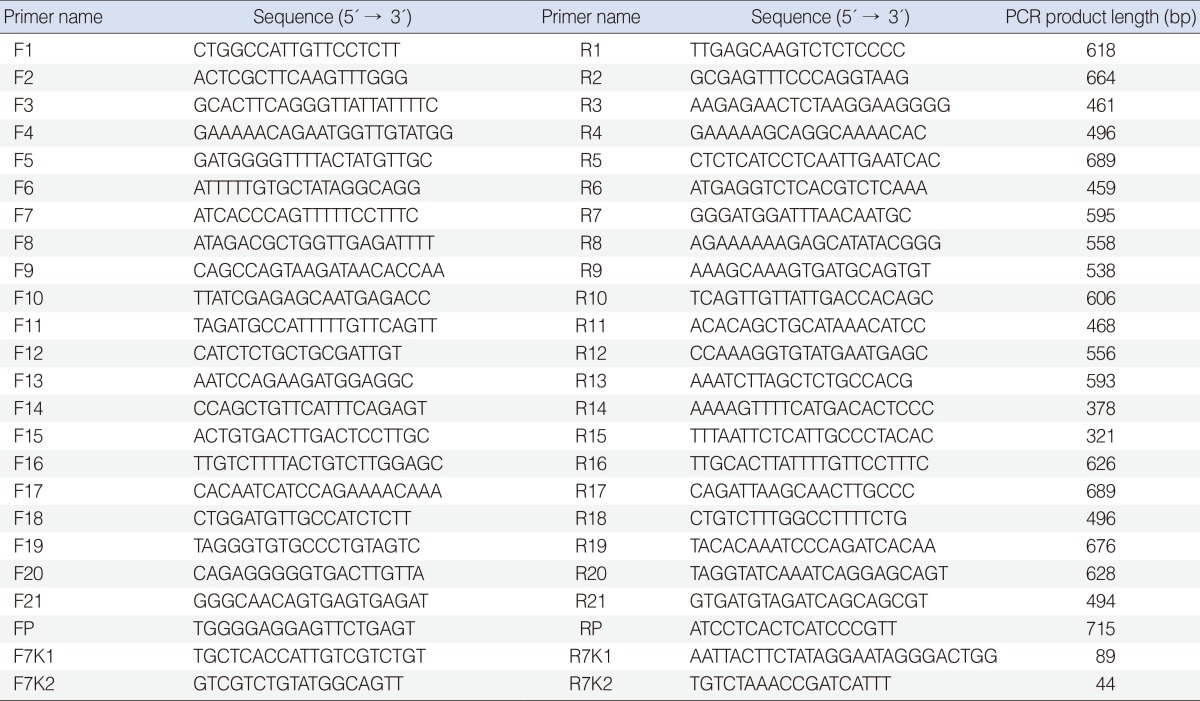
All 21 exons (including 50-200 bp flanking regions) and promoter of SLC26A4 were amplified using primers designed by Oligo ver. 6.7.1.0 (National Biosciences Inc., Plymouth, MN, USA) (Table 1).
Amplification was performed, with some modifications for each amplicons, in a 50 µL volume of reaction, containing 4 µL of MgCl2 (50 Mm), 5 µL of Taq PCR buffer (10X), 0.6 µL of each primer (10 pM), 1 µL of dNTP mix (10 mM), 0.2 µL Taq DNA pol (5 U/µL), 4 µL DNA (50 ng), 34.6 of ddH20. PCR was done according to the following program: an initial denaturation at 95℃ for 5 minutes, 36 cycles of 95℃ for 1 minute (denaturation), 61℃ for 1 minute (annealing), 72℃ for 1 minute (extension) and a final extension at 72℃ for 8 minutes. DNA sequencing of the PCR-amplified product was performed bi-directionally on an ABI 3730XL automated sequencer (Applied Biosystems, Macrogen, Seoul, Korea) using the same primers for amplification.
Pathogenicity investigation of novel variants
Mutation confirmation was done with co-segregation study of the novel variants and their absence in 100 ethnically matched normal control subjects. For investigation of c.881-882delAC variant pathogenicity, a 89 bp fragment of exon 7 harbouring c.881-882delAC location was amplified with primers 7K1F and 7K1R. For studying c.863-864insT pathogenicity, a 44 bp fragment of exon 7, containing the location of this variant, was amplified using 7K2F and 7K2R primers. 7K1 and 7K2 primers were designed by primer3 ver. 0.4.0 (http://frodo.wi.mit.edu/primer3/) and Oligo ver. 6.7.1.0 (National Biosciences Inc.), respectively (Table 1). PCR amplifications were performed using the conditions mentioned above. Amplification products were run on 14% PAGE at 28 mA for 3 hours. Allelic variant c.881-882delAC is 2 bp shorter than wild type alleles and c.863-864insT is 1bp longer than normal type alleles.
RESULTS
Phenotype evaluation results
Audiological testing results
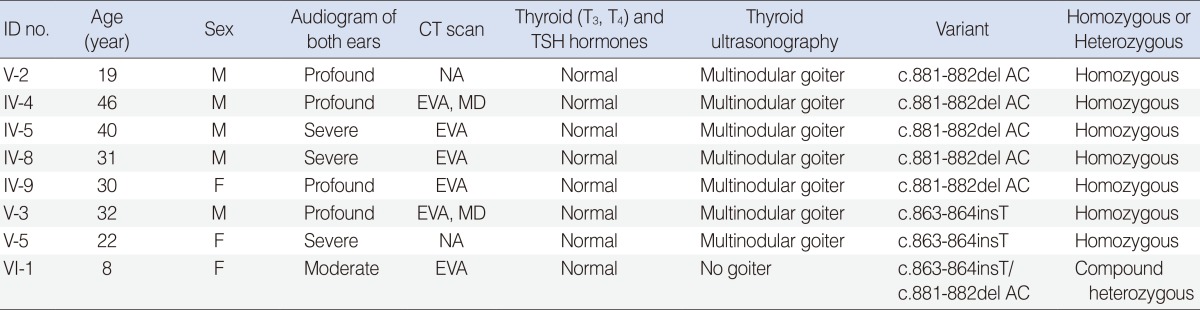
Audiological characterization of all patients is shown in Fig. 2. Mean PTA of thresholds for air conduction at frequencies 500, 1,000, 2,000 Hz was 97.22±7.09 for part A, 90.83±5.20 for part B, 60±5 for patient VI-1 (right ear) and 95.83±5 for part A, 86.67±5.20 for part B and 46.67±2.89 for patient VI-1 (left ear). The severity of HL for all patients has been shown in Table 2.
Molecular studies results
Slink calculation, DFNB4 STR marker genotyping and linkage analysis
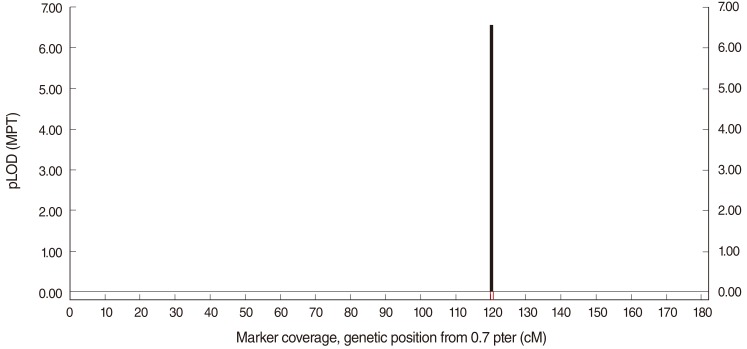
Slink, two-point and multi-point LOD scores were respectively as follows: 4.90, 5.17, and 6.57 (Fig. 5). LOD score >3 confirmed linkage to DFNB4.
Mutation screening of SLC26A4 results
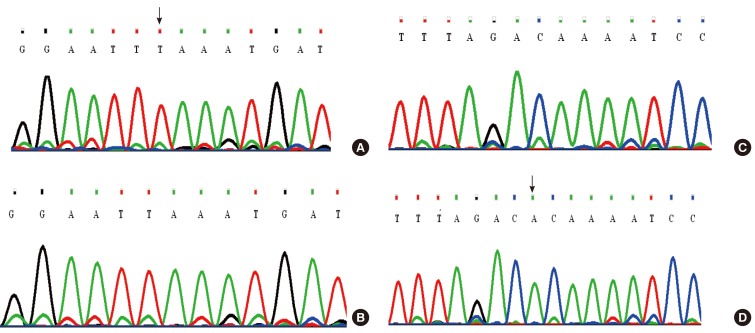
Finding of two different homozygous haplotypes in the patients of part A and part B of the pedigree and the simultaneous presence of both haplotypes in patient V1-1 predicted possible compound heterozygosity (Fig. 1), which was later confirmed by DNA sequencing: two novel variants including c.863-864insT (p.Leu-288PhefsX3) and c.881-882delAC (P.His294GlnfsX35) were identified in exon 7 of SLC26A4 (Fig. 6). Homozygous c.863-864insT variant was found in Patients V-3 and V-5, while subjects V-4, V-7, IV-20, IV-19, IV-18, IV-17, IV-13, and IV-10 were heterozygous for this variant. Individuals II-4, III-1, III-2, III-3, IV-1, IV-2, and IV-3 were heterozygous for c.881-882delAC. Patients IV-4, IV-5, IV-8, IV-9, V-1, and V-2 were homozygous for this variant. Compound heterozygosity for both of these variants was revealed in patient VI-1.
Pathogenicity confirmation of novel variants
The variants c.881-882delAC and c.863-864insT were found in a homozygous state in patients of the pedigree but were not detected, or were in heterozygous state, in normal subjects of the family. None of these variants were detected in 100 ethnically matched normal control subjects. With respect to these results and the nature of the two variants (frameshift), we suggest the pathogenicity role of both variants.
DISCUSSION
In the present study, we detected two novel variants (c.863-864insT and c.881-882delAC) in a large consanguineous Iranian family with 9 patients with HL linked to DFNB4 locus. c.863-864insT with a frameshift at codon 288 causes a premature stop at position 290 and c.881-882delAC results in a frameshift at codon 294 and a premature stop at position 328.
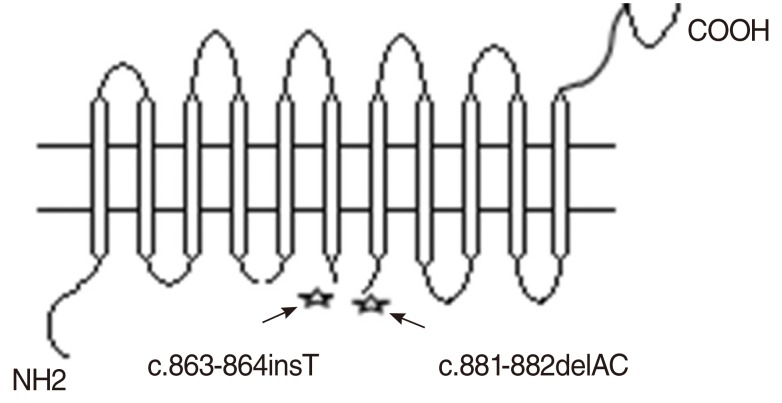
There is no definite structure for pendrin but a few models have been predicted for this protein. Based on pendrin structural modeling, predicted by Everett et al. [3] and the model described in Pendred/Bor homepage (http://www.healthcare.edu/labs/pendredandbor/domains.htm), c.863-864insT and c.881-882delAC lead to amino acid changes at intracellular region between the sixth and seventh transmembrane domains of the protein (Fig. 7). Thus, these variants can delete more than half of pendrin and cause a defective protein without a long C terminal segment which probably is not processed to reach the plasma membrane and would not be functional.
However, further molecular studies on mRNA expression might be necessary to help gain a more comprehensive understanding of the exact effect of these two novel variants on structure and function of the resulting protein.
In this study, patient VI-1, aged 8 years old, had normal thyroid size and function and a moderate HL. She was compound heterozygous for c.863-864insT and c.881-882delAC. The combined presence of the two variants may lead to a different phenotype from that caused by any single variant alone in the homozygous state. Alternatively, PS-related phenotypes in the patient might be progressive and could be more severe with age. As goiter does not usually manifest until adulthood and even after that, the patient VI-1 may show goiter at an advanced age. Furthermore, according to Scott et al. [21] study, PS and DFNB4 SLC26A4 mutations are different functionally. Mutations, leading to PS are usually associated with no transporting ability, but those accounting for DFNB4 HL may show some residual iodide and chloride transporting function.
However, no obvious relationship has been reported between SLC26A4 mutations and phenotype by some investigators [22, 23]. These contradictory results could suggest that some other modifier genes, epigenetic and environmental factors such as nutritional iodide uptake can influence the clinical manifestations due to SLC26A4 mutations. Other patients in the pedigree had mostly severe to profound HL, and were positive for EVA and euthyroid goiter. These results show c.863-864insT and c.881-882delAC would most probably cause PS with rather similar phenotypic patterns.
It has been suggested that SLC26A4 would play a more considerable role in HI of Asian populations, such as Iranians, than other populations [11-15,24]. Up to now a considerable number of all mutations, which have been detected in Iranian HL patients have been novel [13,25,26]. In the present study, we detected two different novel variants segregating with HL in a large deaf family. Taken together, these different studies probably confirm the high frequency and specificity of SLC26A4 mutations and the critical role of these defects in causing HI in Iranian heterogenous population. Therefore it is beneficial to consider screening of SLC26A4 mutations in molecular diagnostics of HL. Furthermore, Detection of mutations in this gene may have implications for better understanding of the exact structure and function of pendrin as well as designing of comfortable and more cost effective strategies in genetic diagnosis of HI.