INTRODUCTION
Enlarged vestibular aqueduct (EVA; OMIM: 600791), the most abundant malformation of the inner ear, is often found by computed tomography (CT) in patients with sensorineural hearing loss (HL) [1]. Nonsyndromic HL with EVA is typically characterized by congenital, bilateral sensorineural HL, which can be progressive and usually ranges from severe to profound [2]. However, patients manifest EVA of various shapes and sizes, so the criteria for defining EVA differ depending on the size and measuring position [3,4]. Okamoto et al. [4] divided EVA cases into subgroups based on the shape and size of the vestibular aqueduct and tested for a relationship between the subgroups and HL characteristics or genotypes. However, differences in EVA manifestations did not correlate with HL levels or genotypes [4].
Mutations in SLC26A4 (OMIM: 605646) are among the most prevalent causes of EVA and are regarded the second most frequent cause of autosomal recessive nonsyndromic sensorineural HL after mutations in GJB2 [5]. To date, more than 170 SLC26A4 mutations have been reported in diverse populations (Pendred/BOR Homepage: http://www.healthcare.uiowa.edu/labs/pendredandbor/slcMutations.htm) as the cause of both nonsyndromic HL with EVA and Pendred syndrome (PS; OMIM: 274600) associated with other clinical findings, such as goiter, Mondini dysplasia, and incomplete iodide organification [6,7]. SLC26A4 comprises 21 exons and is situated on chromosome 7q31 [6]. Pendrin, the gene product of SLC26A4, is a 780-amino acids hydrophobic membrane protein with 12 transmembrane domains [6,7]. Pendrin plays important roles in fluid and anion transport and maintenance of the endocochlear potential by mediating the exchange of bicarbonate, formate, chloride, and iodide ions [7]. Mutations in SLC26A4 can result in anion transport impairments in the thyroid and inner ear causing HL and/or goiter [8].
In this study, we describe a Korean family with HL, including young siblings with sensorineural HL with EVA and compound heterozygous mutations in the SLC26A4 gene.
MATERIALS AND METHODS
Subjects and clinical evaluations
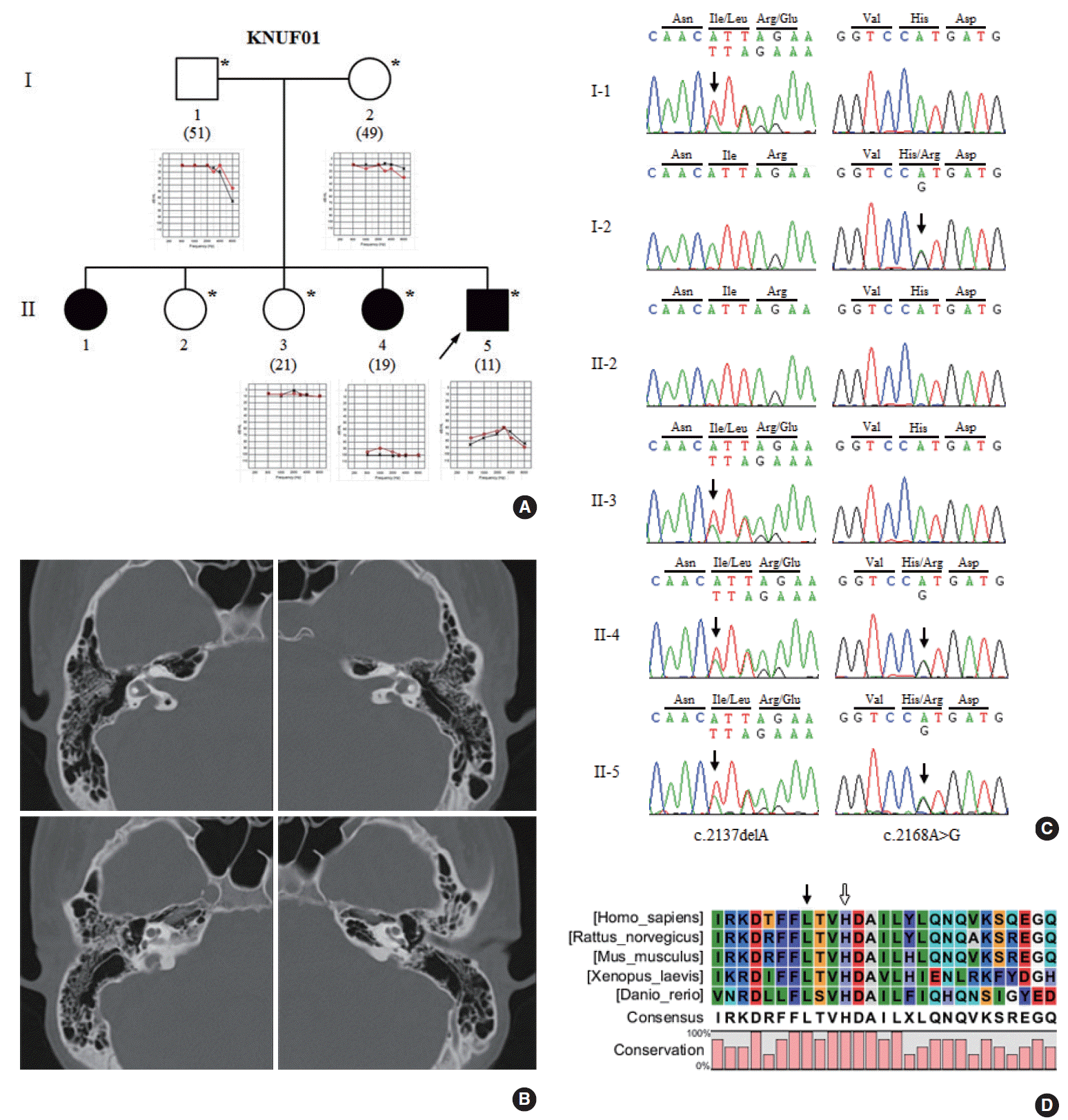
A Korean family with HL (KNUF01) was recruited from the Department of Otorhinolaryngology-Head and Neck Surgery at the Kyungpook National University Hospital in Daegu, Korea. Clinical evaluation of the family was conducted including the description of the family history, medical anamneses, and audiological testing for hearing level complemented by pure-tone audiometry (PTA) (Fig. 1A). We performed PTA with air-conduction at 500ŌĆō8,000 Hz and computed the mean of thresholds checked at 500, 1,000, 2,000, and 4,000 Hz. The degree of HL was estimated based on PTA results as previously described [9]. Temporal bone CT was performed using a Somatom Sensation 16 (Siemens, Erlangen, Germany). One hundred unrelated Koreans who underwent PTA testing at the Kyungpook National University Hospital were recruited as normal controls. We obtained written informed consent from all participants, and this study was approved by the Institutional Review Board of the Kyungpook National University Hospital (KNUH BIO_09-1007).
Mutation analysis
A FlexiGene DNA kit (Qiagen, Hilden, Germany) was used to extract genomic DNA from the peripheral blood of the KNUF01 family members and 100 normal controls. All 21 exons and the exonŌĆōintron boundaries of SLC26A4 (NM_000441.1) were analyzed by PCR using followed primer sets designed with the Primer3 software (http://primer3.ut.ee/) (Table 1), and their nucleotide sequences were analyzed using direct sequencing as described previously [10]. Mutation analysis was performed using a 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA), Sequencing Analysis ver. 5.2 (Applied Biosystems), and Chromas Lite ver. 2.01 (Technelysium, Tewantin, QLD, Australia). The 1000 Genomes Project database (http://www.1000genomes.org/) and the dbSNP database of the National Center for Biotechnology Information (NCBI; http://www.ncbi.nlm.nih.gov/) were used as references to assess the novelty of mutations found in this study. The potential pathogenicity of the novel mutation was predicted by MutationTaster (http://www.mutationtaster.org/), and multiple sequence alignments using CLC Sequence Viewer ver. 6.0.1 (CLC Bio, Aarhus, Denmark) were carried out to investigate the evolutionary conservation of the mutated amino acids.
RESULTS
Clinical evaluation
An 11-year-old boy (II-5) complained of HL and was examined for hearing level. His family (KNUF01) displayed autosomal recessive transmission (Fig. 1A). As the result of PTA, II-5 showed moderately severe HL with 70 and 73.3 dB average air-conduction in the right and left ears, respectively. As a result of periodic examination, he had revealed fluctuating HL at the age of seven. His sister (II-4), another affected family member, had exhibited profound bilateral HL from 5 years old. Other family members (I-1, I-2, and II-3) showed normal hearing. However, the affected sibling (II-1) was not available for study. Temporal bone CT of the proband showed Mondini dysplasia and bilateral EVA with width of the vestibular aqueduct greater than 1.5 mm [11] (Fig. 1B). Thyroid functional tests revealed normal serum concentrations of relevant substances: free thyroxine, 1.2 ng/dL; triiodothyronine, 1.6 ng/mL; thyroid-stimulating hormone, 2.32 ╬╝IU/mL; and antithyroglobulin antibody, 21.5 U/mL. His neck ultrasonography produced normal results and did not reveal any pathologic lesions.
Mutation analysis
We identified two SLC26A4 variations in this family. The first variation was deletion of an adenine (c.2137delA) in exon 19 (left panels in Fig. 1C). This novel deletion was predicted to cause a frameshift and produce a truncated protein by a premature stop (p.I713LfsX8). Four members (I-1, II-3, II-4, and II-5) of the KNUF01 family were heterozygous for this variation. However, it was not detected in two other family members (I-2 and II-2) or in 100 unrelated controls. One member (II-1) of this family was not available for this study. This variation was not reported in the NCBI dbSNP or 1000 Genomes Project databases, and truncated region caused by this variation was located in a highly conserved region among mammals (Fig. 1D). In addition, MutationTaster predicted this variation to be disease causing.
The second mutation was an adenine to guanine substitution (c.2168A>G) in exon 19 (right panels in Fig. 1C), which leads to substitution of histidine by arginine (p.H723R). Three members (I-2, II-4, and II-5) of the KNUF01 family were heterozygous for this mutation. This variation was present in the NCBI dbSNP and 1000 Genomes Project databases with a minor allele frequency (MAF) of 0.04% (rs121908362). In addition, this variation was previously reported to cause PS or nonsyndromic HL with EVA. The affected members (II-4 and II-5) of the KNUF01 family were heterozygous for both p.I713LfsX8 and p.H723R mutations. This finding indicates that the novel p.I713LfsX8 variation is the disease-causing mutation in this family as a compound heterozygous mutation with p.H723R.
DISCUSSION
Compound heterozygous patients with two SLC26A4 mutant alleles comprised up to 50% of Korean and Chinese patients and 70% of Japanese patients with HL [12,13]. We also identified compound heterozygous mutations with p.H723R and p.I713LfsX8 in two individuals in a family. These mutations were both located in exon 19 of SLC26A4. This exon is a mutation hotspot in East Asian populations and is particularly frequently mutated in the Korean population (Fig. 2)[12-22]. Among previously described mutant alleles in exon 19, the p.H723R mutation identified in this study was the most common mutation among Korean and Japanese patients with HL [12-15,17,18]. Yoon et al. [23] reported that the pendrin produced with this mutation was localized in endoplasmic reticulum tube-like structures and lacked ClŌĆō/HCO3ŌĆō exchange activity, whereas wild-type proteins reached the plasma membrane.
The other mutation, p.I713LfsX8, was reported for the first time in this study. This mutation led to early translational termination at amino acid position 720 in the sulfate transporter and anti-sigma factor antagonist (STAS) domain, which also modified the putative protein kinase A (PKA) binding site. The PKA site and the distal part of the STAS domain play an important role in regulating pendrin localization to the plasma membrane. Bizhanova et al. [24] reported that phosphorylation of the putative PKA site is not an absolute prerequisite for pendrin targeting to the plasma membrane. However, the STAS domain included in members of the SLC26A family regulates the stability, trafficking, and anion transport function of SLC26 family proteins [7,25]. The structural significance of this domain has been substantiated by the disease-causing nature of mutations therein among SLC26A family proteins [25]. Therefore, it is possible that the novel mutation discovered in our study impairs the function of pendrin by inactivating the distal STAS domain. Additionally, early stop codon causes nonsense-mediated mRNA decay (NMD) and nonsense mRNAs are rapidly decayed [26]. Therefore, the concentration of truncated protein would be extremely low in vivo. Even if the truncated protein is expressed, loss of the important functional domains will decide the abnormal levels of proteins. Because the patient had both p.I713LfsX8 and p.H723R mutations in each allele, another full-length proteins carrying the p.H723R mutation are also abnormal. Therefore, the patient cannot have any normal pendrin proteins.
Mutations in SLC26A4 have been reported to be responsible for a wide phenotypic spectrum of hearing problems, from typical PS to nonsyndromic HL with EVA [1,6]. There have been many reports documenting genotype-phenotype correlations in patients with mutations in SLC26A4. Several studies reported that patients with bi-allelic SLC26A4 mutations showed more severe HL, larger malformations of the inner ear, and more pronounced goiter than patients with no or mono-allelic mutations [27,28]. On the other hand, other studies suggested that there was no correlation between HL and SLC26A4 mutations [19,29]. The patient with bi-allelic mutations examined in this study showed profound HL but no evidence of goiter.
In conclusion, we describe here a case of nonsyndromic HL with EVA in young siblings of a Korean family, whose members harbored mutations in SLC26A4. We found compound heterozygous mutation of the SLC26A4 gene including p.I713LfsX8, a novel frameshift mutation. Thus, our study could provide a foundation for future investigation of the mechanisms of SLC26A4-associated HL.